CHARMM-GUI Ligand Binder for Absolute Binding Free Energy Calculations and Its Application
By Sunhwan Jo, Wei Jiang, Hui Sun Lee, Benoı̂t Roux, and Wonpil Im.
Published in Journal of Chemical Information and Modeling 53(1): 267-277 (2013) on January 28, 2013. PMID: 23205773. PMCID: PMC3557591. Link to Pubmed page.
Core Facility: Computational Modeling
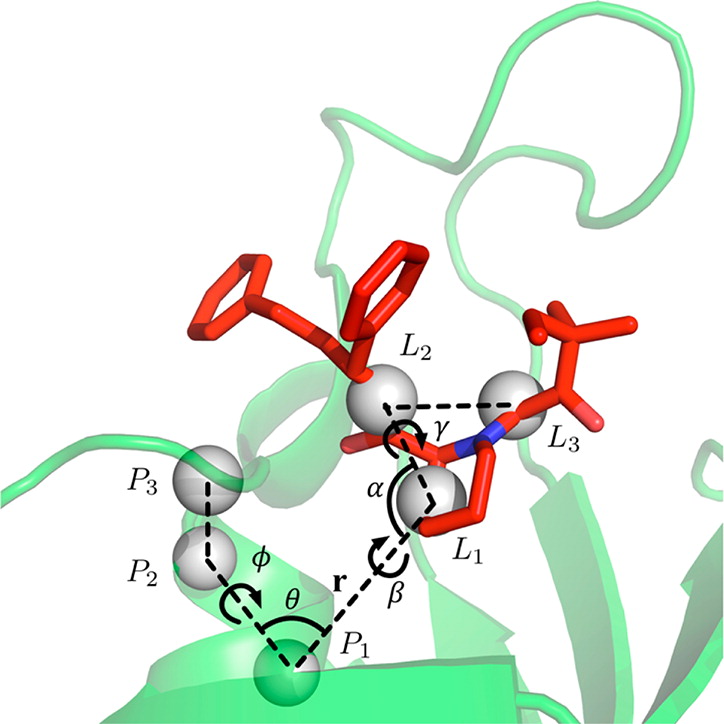
Figure 1. Illustration of translational and orientational restraints on SB3 ligand in PDB:1FKG (see also Figure 2). Anchoring atoms are represented as gray sphere. r (P1-L1), θ (P2P1L1), (P3P2P1L1) are defined for the translational restraint, and α (P1L1L2), β (P2P1L1L2), and γ (P1L1L2L3) are defined for the orientational restraint.
Abstract
Advanced free energy perturbation molecular dynamics (FEP/MD) simulation methods are available to accurately calculate absolute binding free energies of protein-ligand complexes. However, these methods rely on several sophisticated command scripts implementing various biasing energy restraints to enhance the convergence of the FEP/MD calculations, which must all be handled properly to yield correct results. Here, we present a user-friendly Web interface, CHARMM-GUI Ligand Binder, to provide standardized CHARMM input files for calculations of absolute binding free energies using the FEP/MD simulations. A number of features are implemented to conveniently set up the FEP/MD simulations in highly customizable manners, thereby permitting an accelerated throughput of this important class of computations while decreasing the possibility of human errors. The interface and a series of input files generated by the interface are tested with illustrative calculations of absolute binding free energies of three nonpolar aromatic ligands to the L99A mutant of T4 lysozyme and three FK506-related ligands to FKBP12. Statistical errors within individual calculations are found to be small (∼1 kcal/mol), and the calculated binding free energies generally agree well with the experimental measurements and the previous computational studies (within ∼2 kcal/mol). Therefore, CHARMM-GUI Ligand Binder provides a convenient and reliable way to set up the ligand binding free energy calculations and can be applicable to pharmaceutically important protein-ligand systems.

 SITE and (B) BULK using the GSBP and SSBP boundary potentials.")
