Investigating Conformational Changes in a Saccharide Transporter
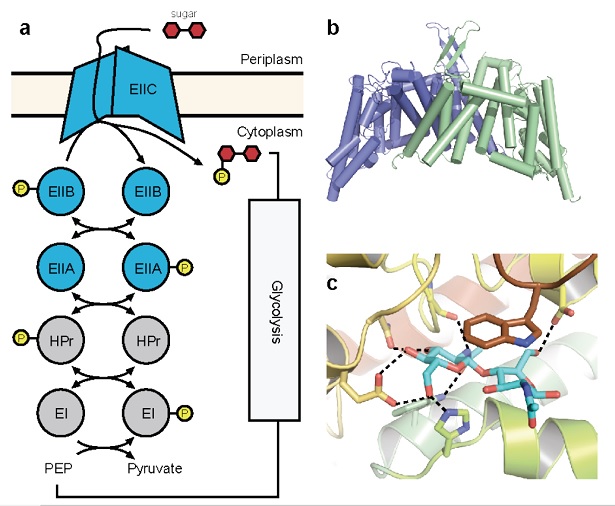
Figure 1: Crystal structure of ChbC. (a) Diagram of components of a generalized PTS system. (b) The ChbC dimer, colored by subunit. (c) The diacetylchitobiose-binding pocket.
Project Description
Transporters are integral membrane proteins that conduct small molecules across lipid bilayers via aseries of conformational changes that alternately expose the permeant molecules to one side of the membrane or the other. Unlike channels, which generally contain continuous pores that assist molecules in traversing down their electrochemical gradients, the alternative-access mechanisms of transporters allow them to couple an energetic driving force to the active transport of a molecule in a thermodynamically unfavorable direction. This ability can serve to concentrate nutrients, expel unwanted compounds, or to store potential energy as an electrochemical gradient for future use. The two best structurally characterized classes of transporters are the primary transporters, which use the chemical energy from ATP hydrolysis to drive active transport, and the secondary transporters, which couple transport to the gradient of a secondary compound.
A third strategy that transport systems can employ to concentrate a compound against its electrochemical gradient is to couple transport with the covalent modification of the permeant molecule. One notable example is the phosphenolpyruvate-dependent phosphotransferase system (PTS), a multicomponent uptake system found only in bacteria that transports sugars favored for metabolism against their concentration gradient into the cell. The energy for transport derives from phosphorylation of the sugar molecule synchronously with translocation across the bilayer. PTSs comprise a series of cytoplasmic phospotransferases and an integral membrane component, EIIC (Figure 1a). A phosphate group contributed by phosphenolpyruvate is shuttled through the system to the final phosphotransferase EIIB, which transfers the phosphate directly to an incoming sugar translocated by EIIC. Although EIIC does not form a covalent adduct with the phosphate group, its presence is required for phosphorylation to proceed, suggesting that EIIC forms part of the active site and giving rise to the observed tight coupling between transport and phosphorylation.
The PTS has been the subject of considerable interest for many decades, both because it is required for the growth of many pathogenic bacteria, and because it is a model system for how bacteria can link regulation of metabolic and other pathways to the nutritive qualities of their environment. Cognate PTS saccharides produced intracellularly by conversion of other sugars are not phosphorylated at appreciable rates, and consequently, the phosphorylation states of PTS components are diagnostic of the availability of the saccharide in the external environment, and are used by the bacterium in signal transduction to regulate pathways relevant to carbohydrate metabolism. The PTS regulates expression of genes involved in cognate sugar uptake, while concurrently repressing pathways for the metabolism of non-PTS sugars, including by directly inhibiting the transporters responsible for their uptake. In addition to regulating sugar metabolism, the PTS is involved in a number of other pathways that are necessary for bacteria to respond appropriately to the presence or absence of nutrients in their environment, including chemotaxis and biofilm formation.
To understand better the structural basis of PTS-mediated transport, we solved the first structure of a PTS EIIC transporter, ChbC, which is specific for the transport of the mono-, di- and tri-saccharide of N-acetylglucosamine (GlcNAc). The structure reveals a ten-transmembrane helix fold with no detectable structural homology with other transporters of known structure (Figure 1b). The ChbC protomer can be divided into two domains: one forming a dimer interface with another ChbC protomer, and binding domain containing a substrate molecule, most likely the (GlcNAc)2 disaccharide, located in a large, electronegative binding cavity near the cytoplasmic side of transporter (Figure 1c). Because there is no pathway from the binding cavity to the cytoplasm large enough to permit the substrate, this conformation likely corresponds to the inward-facing occluded state of the transporter. Based on this crystal structure, we have proposed a tentative mechanism for how the sugar is translocated across the bilayer (Figure 2). Experimental confirmation of this mechanism, however, represents a considerable technical challenge, as devising a strategy to trap a particular conformation in a crystal for diffraction studies is not straightforward. Here we propose a series of experiments to complement our crystallographic studies of ChbC-mediated phosphorylation-coupled transport by using double electron-electron resonance (DEER) electron paramagnetic resonance (EPR) spectroscopy and molecular dynamics simulations to probe the conformational changes and energetics underlying the transport cycle of ChbC.
I. Conformation of the outward-open state.
The crystal structure of ChbC reveals a buried, solvent-inaccessible substrate-binding cavity, and most likely corresponds to an inward-occluded state in the transport cycle (Figure 2, center). In order to bind substrate from the periplasm, the transporter must possess an alternate structure with the binding cavity exposed to the periplasmic side, but the crystal structure provides few clues as to how this conformational change might occur. The first goal of this proposal is to test and refine our model for the conformation of the outward-open state, and to identify specific interactions with the ligand that affect the equilibrium between this and the inward-occluded state observed in the crystal structure. Our model of the outward-open state predicts that a region of the protein comprising helices TM8-10 and the two hairpin loops HP1 and HP2 undergoes a rigid body motion relative to the immobile dimer interface domain (Figure 2, left). To test this theory, we will introduce pairs of cysteine mutations to residues located on the periplasmic-facing ends of TM1, TM7, HP2 and TM8, in order to attach spin labels. We propose to then reconstitute the labeled proteins into bilayers and use DEER EPR to measure the distribution of distances between the ends of TM8 and HP2 on the predicted mobile domain and TM1 and TM7 on the immobile interface domain in the presence and absence of ligand. If the current model is correct, the distributions should shift from long distances between the labels (corresponding to the outward-open state) to shorter distances matching the crystal structure (the inward-occluded state) when substrate is bound. Once we have identified intramolecular distances characteristic of the outward and inward-facing states, we will design mutations aimed at understanding how interactions with the substrate shift the conformational equilibrium of the protein from one state to another.
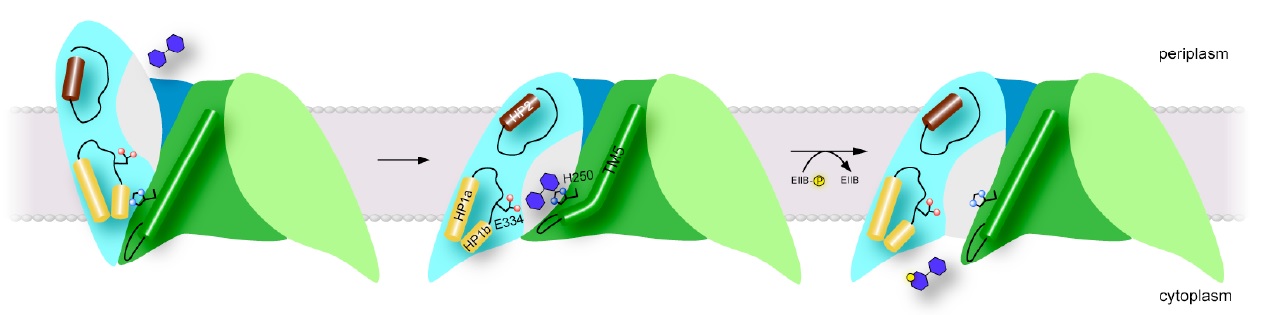
Figure 2: Proposed transport mechanism. Schematic of proposed models for the outward-open (left), occluded (center) and inward-open states (right) of ChbC. Click to enlarge.
II. Conformation of the inward-open state.
While the substrate in the ChbC crystal structure is located close to the intracellular side, the binding pocket is still occluded from solution, primarily by the loop between helices TM4 and TM5. Our current hypothesis is that the TM4-5 loop acts as an intracellular gate, and substrate is released by a swinging movement of this loop facilitated by a straightening of a kink in TM5, exposing the binding site to the cytoplasm (Figure 2, right). The tight coupling between phosphorylation and substrate release observed in cells suggests that the conformational change to release substrate is likely accelerated by binding to ChbB and the transfer of the phosphate to the substrate. However, even in the absence of ChbB, we were able to observe slow passive transport of the ligand across bilayers mediated by ChbC, indicating that the conformational change to the inward-open state can still occur, albeit at a reduced rate, without formation of the ternary complex between ChbB, ChbC, and the substrate.
The unliganded protein, in order to be competent to bind substrate from the periplasm, is expected to assume the outward-open conformation. We therefore expect that the inward-open conformation is a transient state along the pathway to revert back to the outward-open conformation after release of substrate. This would make solving the structure of the inward-open conformation by X-ray crystallography extremely challenging. However, the proposed movement of the TM4-5 loop is a small enough conformational change to be potentially observable on the timescales feasible for unbiased molecular dynamics simulations. We therefore propose to test our model for the inward-open state and release of the substrate by carrying out molecule dynamics simulations on (GlcNAc)2-bound, unliganded and (GlcNAc)2-PO3 bound ChbC. While release of unphosphorylated (GlcNAc)2 is likely too slow occur on the microsecond timescales accessible to molecular dynamics simulations, repulsion between the phosphate group and the electronegative binding pocket likely increases the rate of gate opening and substrate release. It is also possible that the loss of interactions with the substrate in the unliganded form would also destabilize the occluded conformation, allowing observation of the opening of the intracellular gate.
The above experiments will complement the aims of our currently active R01 (R01GM098878), which proposes to solve crystal structures of the outward-open conformation of ChbC and of the complex with ChbB. In particular, the DEER EPR experiments described in Aim 1 may result in the identification of mutations that shift the structural equilibrium of the protein towards the outward-open state. These mutation could be combined be produce a variant protein that will remain in the outward-open conformation irrespective of the influences of detergent or the crystal lattice, increasing the chances of crystallizing and solving the structure of that state. Similarly, an accurate model of the inward-open state derived from the molecular dynamics simulations from Aim 2 could improve our ability to design a stabilized ChbB/ChbC/substrate complex for crystallization.
