xMDFF: molecular dynamics flexible fitting of low-resolution X-ray structures
By Ryan McGreevy, Abhishek Singharoy, Qufei Li, Jingfen Zhang, Dong Xu, Eduardo Perozo, and Klaus Schulten.
Published in Acta Crystallogr D Biol Crystallogr. 2014 Sep;70(Pt 9):2344-55. PMID: 25195748. PMCID: PMC4157446. Link to publication page.
Core Facilities: Membrane Protein Expression and Purification and Computational Modeling.
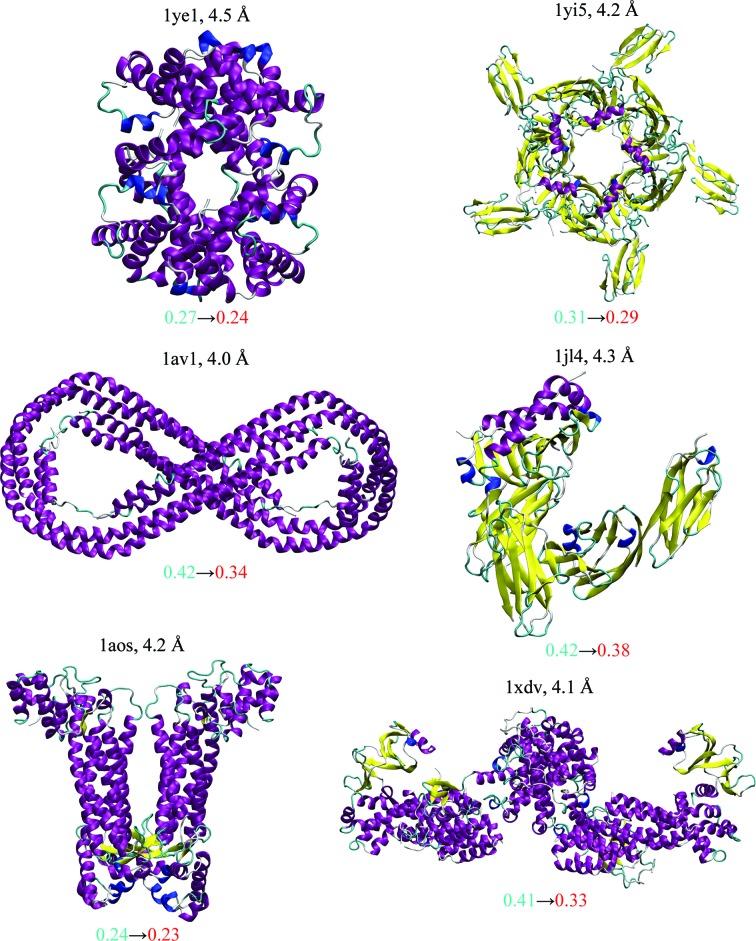
Figure 1. xMDFF re-refinement characteristics for six structures. The structures with diffraction data between 4 and 4.5 Å resolution had been deposited earlier in the Protein Data Bank and were used as initial models for xMDFF refinement. Improvements were evaluated using the R free values of the xMDFF-refined structures (shown in red) against their respective starting values taken from the deposited structure (blue). In addition, the increase in the percentage of favored Ramachandran angles owing to xMDFF refinement and other structural statistics, as summarized by the overall MolProbity score, were used to measure the improvement in structural geometries.
Abstract
X-ray crystallography remains the most dominant method for solving atomic structures. However, for relatively large systems, the availability of only medium-to-low-resolution diffraction data often limits the determination of all-atom details. A new molecular dynamics flexible fitting (MDFF)-based approach, xMDFF, for determining structures from such low-resolution crystallographic data is reported. xMDFF employs a real-space refinement scheme that flexibly fits atomic models into an iteratively updating electron-density map. It addresses significant large-scale deformations of the initial model to fit the low-resolution density, as tested with synthetic low-resolution maps of d-ribose-binding protein. xMDFF has been successfully applied to re-refine six low-resolution protein structures of varying sizes that had already been submitted to the Protein Data Bank. Finally, via systematic refinement of a series of data from 3.6 to 7 Å resolution, xMDFF refinements together with electrophysiology experiments were used to validate the first all-atom structure of the voltage-sensing protein Ci-VSP.
. Biasing forces f fit derived from a Φ(r)-dependent potential are used to flexibly fit the phasing model into the map, yielding structures most consistent with the electron density. With the most recently fitted structure as a new phasing model, density maps are synthesized and flexibly fitted until a structure is found with sufficiently low R free, providing the best possible interpretation of the diffraction data.")
 of the d-ribose-binding protein (a) and diffraction data from the target closed structure (PDB entry 2dri; orange) at resolutions ranging from 3.5 to 5 Å, xMDFF refinements were performed. (b) The xMDFF-refined closed structure (red) shows excellent agreement with the target at all resolutions (5 Å resolution shown with a final R free of 0.34, down from an initial 0.56). Overall improvement was characterized using the R free and cross-correlation of the final xMDFF structure and the r.m.s.d. of the final xMDFF structure to the target. Structural refinement is suggested by all three measures as well as the all-atom statistics provided in Table 1")
 (a) and xMDFF-refined final (red) (b) structures; local cross-correlations increase from 0.47 to 0.63, implying a more unambiguous placement of the atoms.")
 A MUFOLD-predicted homology model (cyan) was used as an initial phasing model in xMDFF; this model has an r.m.s.d. of 6 Å from an independently refined Ci-VSP structure (orange; Li et al., 2014 [triangle]). Ci-VSP includes a transmembrane (TM) domain and was crystallized with an antibody (Ab). Inset: the placement of the S4 helix within the TM region which determines the voltage-gating capabilities of the protein is of particular interest. (b) xMDFF refinement with 4 Å resolution diffraction data produced a final structure (red) 2.6 Å away from the independently refined Ci-VSP structure (orange). Inset: the placement of the S4 helix in the Down position and the alignment of the regularly placed voltage-sensing Arg residues is in agreement with the independently refined model (Li et al., 2014 [triangle]).")
