Atomic basis for therapeutic activation of neuronal potassium channels
By Robin Y. Kim, Michael C. Yau, Jason D. Galpin, Guiscard Seebohm, Christopher A Ahern, Stephen Pless and Harley Kutara.
Published in Nature Communications 2915 Sep 3;6:8116.
PMID: 26333338. PMCID: PMC4561856. Link to Pubmed page.
Core Facility: Membrane Protein Expression/Purification.
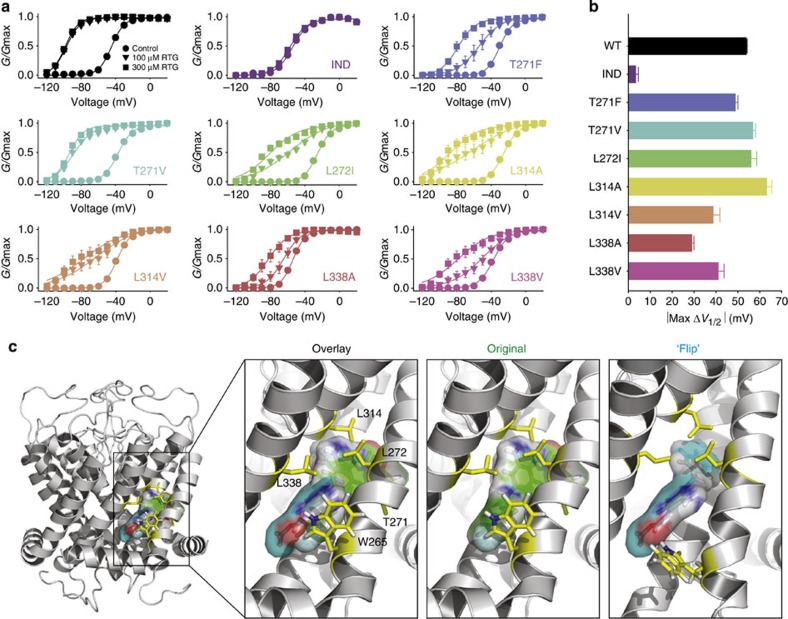
Figure 5. Detailed characterization of secondary retigabine binding residues and alternative binding site orientations. (a) Conductance–voltage relationships were gathered for the indicated KCNQ3* mutant channels (n=4–6 per mutant), in 0, 100 or 300 μM retigabine. (b) Maximal ΔV1/2 in 300 μM retigabine measured in each mutant channel. Error bars in a,b represent s.e.m. (c) Retigabine was docked into a molecular model of the pore-forming domain of KCNQ3 (see ref. 19). Two orientations are shown with the carbamate group in either the vicinity of Leu314 (‘original’ model) or Trp265 (‘flip’ model). The two binding models are superimposed in the ‘overlay’, showing the similar space occupied by both drug orientations.
Abstract
Retigabine is a recently approved anticonvulsant that acts by potentiating neuronal M-current generated by KCNQ2–5 channels, interacting with a conserved Trp residue in the channel pore domain. Using unnatural amino-acid mutagenesis, we subtly altered the properties of this Trp to reveal specific chemical interactions required for retigabine action. Introduction of a non-natural isosteric H-bond-deficient Trp analogue abolishes channel potentiation, indicating that retigabine effects rely strongly on formation of a H-bond with the conserved pore Trp. Supporting this model, substitution with fluorinated Trp analogues, with increased H-bonding propensity, strengthens retigabine potency. In addition, potency of numerous retigabine analogues correlates with the negative electrostatic surface potential of a carbonyl/carbamate oxygen atom present in most KCNQ activators. These findings functionally pinpoint an atomic-scale interaction essential for effects of retigabine and provide stringent constraints that may guide rational improvement of the emerging drug class of KCNQ channel activators.
 Conductance–voltage relationships for (a) KCNQ2 (n=3) and KCNQ2[Trp236Phe] (n=6), and (b) KCNQ3* (n=5) and KCNQ3*[Trp265Phe] (n=3) homomeric channels along with indicated mutants (retigabine concentration of 100 μM). (c) Conductance–voltage relationships for heteromeric combinations of KCNQ2 and KCNQ3 (1:1 ratio of injected mRNA, with or without Trp→Phe mutations as indicated, n=5 for each combination), used to generate channels with reduced numbers of retigabine binding sites. (d) Summary of V1/2 shifts in saturating 100 μM retigabine for mutations of KCNQ2 Trp236 and KCNQ3 Trp265 as indicated (*P<0.05 in a paired Students t-test comparing control versus 100 μM retigabine in each experimental oocyte, n=3–6 per mutant). Only a Trp at either position is sufficient for retigabine sensitivity. (e) Exemplar currents of KCNQ3* and KCNQ3*[Trp265Phe] mutant coexpressed with CiVSP, illustrating that the Trp side chain responsible for retigabine sensitivity is not required for PIP2 sensitivity. (f) Summary data of tail current magnitude (−20 mV) after prepulses to a range of voltages, in oocytes expressing KCNQ3* (n=5) or KCNQ3*[Trp265Phe] (n=5) channels, along with CiVSP. In all panels, error bars represent s.e.m.")
 retigabine, (b) ztz-240 (described in ref. 24), (c) acrylamide (s)-1, (d) BMS-204352 and (e) an unnamed experimental drug described in ref. 43.")
