Determination of Membrane-Insertion Free Energies by Molecular Dynamics Simulations
By James Gumbart and Benoît Roux.
Published in Biophysical Journal 102(4): 795-801 on February 22, 2012. PMID: 22385850. PMCID: PMC3283816. Link to Pubmed page.
Core Facility: Computational Modeling
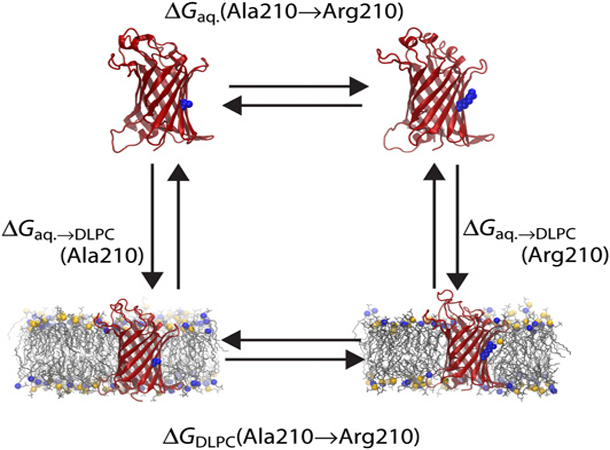
Figure 1.Thermodynamic cycle relating membrane-insertion (vertical legs) with FEP calculations (horizontal legs) for arginine. OmpLA is shown as ribbons and Ala/Arg^210 is indicated at its center in a space-filling representation. The membrane is displayed as thin gray lines with phosphorusatoms of the headgroups as spheres.
Abstract
The accurate prediction of membrane-insertion probability for arbitrary protein sequences is a critical challenge to identifying membrane proteins and determining their folded structures. Although algorithms based on sequence statistics have had moderate success, a complete understanding of the energetic factors that drive the insertion of membrane proteins is essential to thoroughly meeting this challenge. In the last few years, numerous attempts to define a free-energy scale for amino-acid insertion have been made, yet disagreement between most experimental and theoretical scales persists. However, for a recently resolved water-to-bilayer scale, it is found that molecular dynamics simulations that carefully mimic the conditions of the experiment can reproduce experimental free energies, even when using the same force field as previous computational studies that were cited as evidence of this disagreement. Therefore, it is suggested that experimental and simulation-based scales can both be accurate and that discrepancies stem from disparities in the microscopic processes being considered rather than methodological errors. Furthermore, these disparities make the development of a single universally applicable membrane-insertion free energy scale difficult.


