Identification of ligand templates using local structure alignment for structure-based drug design
By Hui Sun Lee and Wonpil Im.
Published in Journal of Chemical Inference and Modeling 52(10): 2784–2795137(10):104101 on September 28, 2012. PMID: 22978550. PMCID: PMC3478504. Link to publication page.
Core Facility: Computational Modeling
Abstract
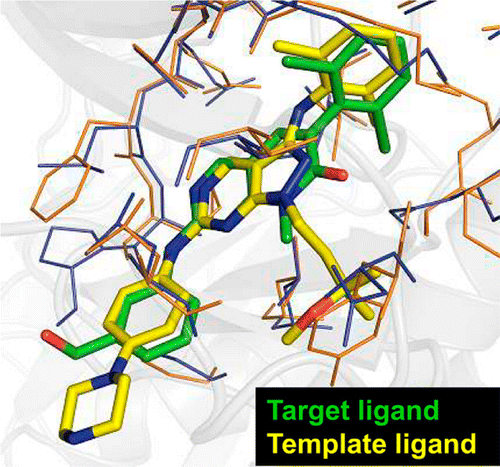
With a rapid increase in the number of high-resolution protein-ligand structures, the known protein-ligand structures can be used to gain insight into ligand-binding modes in a target protein. On the basis of the fact that the structurally similar binding sites share information about their ligands, we have developed a local structure alignment tool, G-LoSA (graph-based local structure alignment). The known protein-ligand binding-site structure library is searched by G-LoSA to detect binding-site structures with similar geometry and physicochemical properties to a query binding-site structure regardless of sequence continuity and protein fold. Then, the ligands in the identified complexes are used as templates (i.e., template ligands) to predict/design a ligand for the target protein. The performance of G-LoSA is validated against 76 benchmark targets from the Astex diverse set. Using the currently available protein-ligand structure library, G-LoSA is able to identify a single template ligand (from a nonhomologous protein complex) that is highly similar to the target ligand in more than half of the benchmark targets. In addition, our benchmark analyses show that an assembly of structural fragments from multiple template ligands with partial similarity to the target ligand can be used to design novel ligand structures specific to the target protein. This study clearly indicates that a template-based ligand modeling has potential for de novo ligand design and can be a complementary approach to the receptor structure based methods.
 is identified. The two local structures are then superposed using a translational and rotational matrix calculated from the identified residue pair set.")
 and template ligands (blue). The template ligand of the best overlap ratio among top 10 templates was used for the structural comparison.")
 and global (TM-align) alignment methods for benchmark target 1HWW and 2BSM (green). The corresponding best templates (blue) were chosen from top 10 templates by G-LoSA.")
 of unsuccessful structure alignment by TM-align-BS between a BS-structure (blue) and a whole structure of its corresponding protein.")
