Potential Application of Alchemical Free Energy Simulations to Discriminate GPCR Ligand Efficacy
By Hui S. Lee, Chaok Seok, and Wonpil Im.
Published in J Chem Theory Comput. 2015 Feb 11;11(3):1255-1266.
Core: Computational Modeling Core.
Abstract
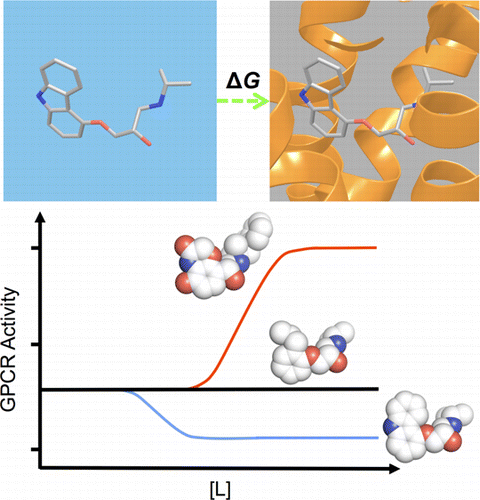
G protein-coupled receptors (GPCRs) play fundamental roles in physiological processes by modulating diverse signaling pathways and thus have been one of the most important drug targets. Based on the fact that GPCR-mediated signaling is modulated in a ligand-specific manner such as agonist, inverse agonist, and neutral antagonist (termed ligand efficacy), quantitative characterization of the ligand efficacy is essential for rational design of selective modulators for GPCR targets. As experimental approaches for this purpose are time-, cost-, and labor-intensive, computational tools that can systematically predict GPCR ligand efficacy can have a big impact on GPCR drug design. Here, we have performed free energy perturbation molecular dynamics simulations to calculate absolute binding free energy of an inverse agonist, a neutral antagonist, and an agonist to β2-adrenergic receptor (β2-AR) active and inactive states, respectively, in explicit lipid bilayers. Relatively short alchemical free energy calculations reveal that both the time-series of the total binding free energy and decomposed energy contributions can be used as relevant physical properties to discriminate β2-AR ligand efficacy. This study illustrates a merit of the current approach over simple, fast docking calculations or highly expensive millisecond-time scale simulations.
 ligands and their binding affinities.")
 SITE and (B) BULK. The β2-AR structure is shown in cartoon representation, the ligand in stick representation, and lipid and water molecules in line representation. K+ and Cl– ions, shown as spheres, in BULK are colored in blue and red, respectively. In SITE, water and ions are not displayed for clarity. Note that the system size of BULK is much smaller than that of SITE, but the figure for BULK here is amplified for clarity.")
 Comparison between I-invC and I-neuC. (B) Comparison between I-invC and I-agoM. The β2-AR residues showing relatively large movement during the activation (>0.8 Å Cα-Cα distance) are colored in red. The distances were measured between β2-AR structures in PDB:2RH1 and PDB:3P0G. In the full agonist figure of (B), the residues that are additionally defined on the basis of I-agoM are also displayed in yellow.")
