Structures of a Na+-coupled, substrate-bound MATE multidrug transporter
By Min Lu, Jindrich Symersky, Martha Radchenko, Akiko Koide, Yi Guo, Rongxin Nie, and Shohei Koide.
Published in Proceedings of the National Academy of Sciences of the United States of America 110(6): 2099-104 (2013) on January 22, 2013. PMID: 22143771. PMCID: PMC3251152. Link to Pubmed page.
Core Facility: Synthetic Antigen Binder (SAB) Generation and Crystallography
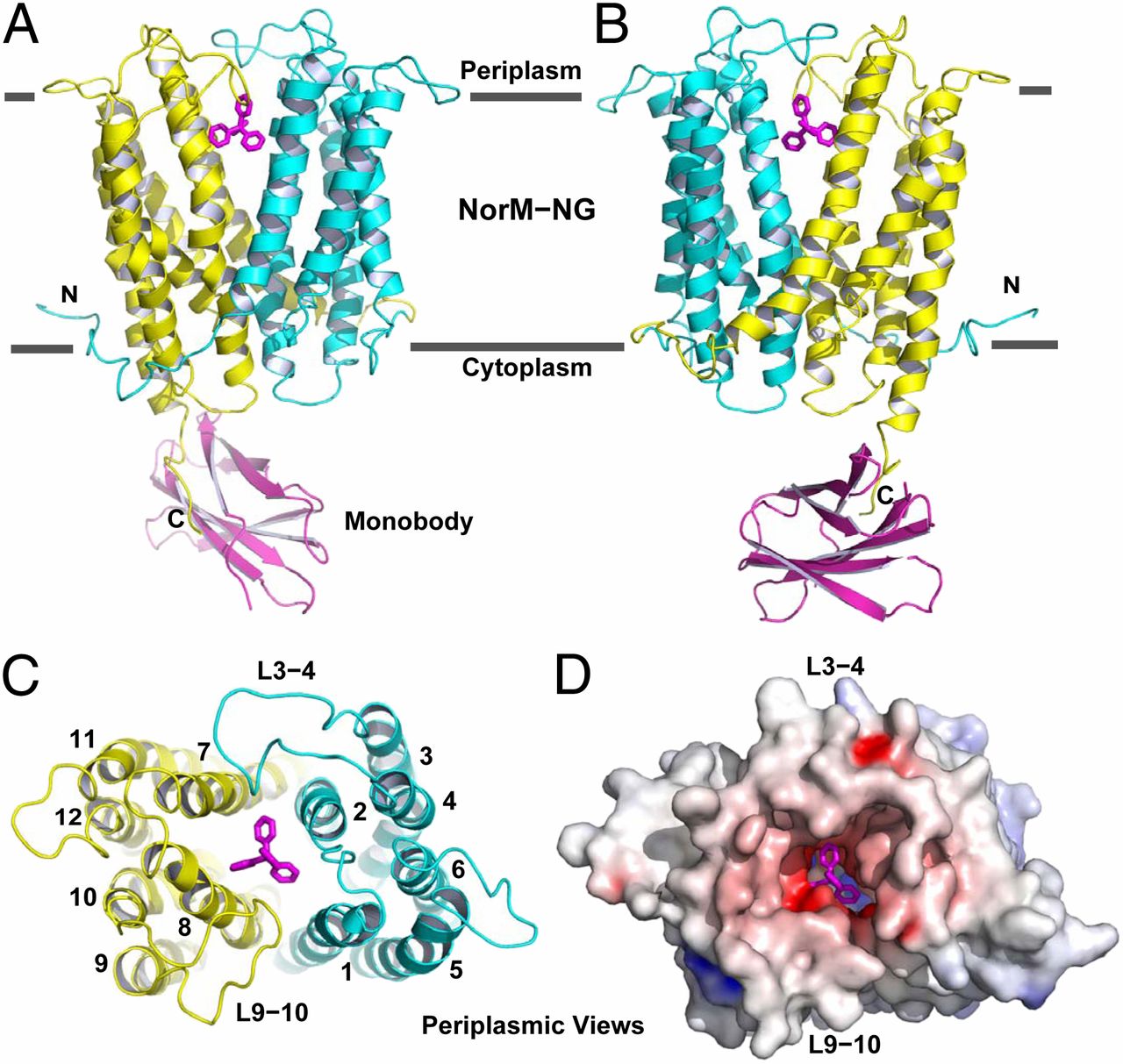
Figure 1. Structure of NorM-NG-monobody complex. (A and B) Structure of NorM-NG monobody complex as viewed from the membrane plane. The views in A and B are related by ∼180° rotation around the membrane normal. The amino (residues 5–230) and carboxyl (residues 231–459) halves of NorM-NG are colored cyan and yellow, respectively. Monobody is shown as a magenta ribbon and bound TPP as magenta sticks. (C) The arrangement of transmembrane helices in NorM-NG as viewed from the periplasmic side. (D) NorM-NG surface as viewed from the periplasmic side, which is colored according to electrostatic potentials from −20 (red) to +20 kTe−1 (blue).
Abstract
Advanced free energy perturbation molecular dynamics (FEP/MD) simulation methods are available to accurately calculate absolute binding free energies of protein-ligand complexes. However, these methods rely on several sophisticated command scripts implementing various biasing energy restraints to enhance the convergence of the FEP/MD calculations, which must all be handled properly to yield correct results. Here, we present a user-friendly Web interface, CHARMM-GUI Ligand Binder ( http://www.charmm-gui.org/input/gbinding ), to provide standardized CHARMM input files for calculations of absolute binding free energies using the FEP/MD simulations. A number of features are implemented to conveniently set up the FEP/MD simulations in highly customizable manners, thereby permitting an accelerated throughput of this important class of computations while decreasing the possibility of human errors. The interface and a series of input files generated by the interface are tested with illustrative calculations of absolute binding free energies of three nonpolar aromatic ligands to the L99A mutant of T4 lysozyme and three FK506-related ligands to FKBP12. Statistical errors within individual calculations are found to be small (∼1 kcal/mol), and the calculated binding free energies generally agree well with the experimental measurements and the previous computational studies (within ∼2 kcal/mol). Therefore, CHARMM-GUI Ligand Binder provides a convenient and reliable way to set up the ligand binding free energy calculations and can be applicable to pharmaceutically important protein-ligand systems.
 NorM-NG is drawn as a ribbon, whereas the substrates are in stick representation. (B–D) Closeup of the binding site for TPP, ethidium (ET), and R6G, respectively. Amino acids within 4.5 Å (given the ∼0.6-Å coordinate errors at current resolutions) of the substrate are illustrated as sticks. L3-4 was omitted in B and C for clarity.")
 Structural overlay of NorM-NG (cyan and yellow, PDB ID code 4HUK) and NorM-VC (gray, PDB ID code 3MKU) (9). TPP (stick model) is colored magenta; red arrows highlight the rearrangement of TM7 and TM8 relative to TM10. (B) Cation-bound structure of NorM-NG (purplish blue ribbon, PDB ID code 4HUL). Cs+ (green sphere) is overlaid with a difference isomorphous Fourier map (magenta mesh) contoured at 6σ. Red arrows indicate proposed movement of TM7 and TM8 toward TM10. TPP (magenta) taken from the TPP-bound structure (PDB 4HUK) is shown in stick representation to indicate the substrate-binding site. (C) Hypothetical Na+ (gray sphere) coordination arrangement that corresponds to state 3 in Fig. 4. Relevant amino acids are depicted as stick models and NorM-NG is colored gray.")
 binds to a cation-free, drug-bound transporter (state 1) and elicits the movement of TM7 and TM8 (red arrow) in the cation-bound, drug-bound protein (state 2), causing the drug to dissociate. The cation-bound, drug-free transporter (state 3) then switches to the inward-facing conformation (state 4), before it binds another drug molecule (magenta). Drug-binding triggers the movement of TM7 and TM8 (red arrow), thereby weakening the Na+ binding (state 5). Na+ releases into the cytoplasm in an inward-facing, drug-bound transporter (state 6), and the transporter returns to the outward-facing, drug-bound conformation (state 1) to complete the transport cycle. Our cation-free, drug-bound NorM-NG structures (PDB ID codes 4HUK, 4HUM, and 4HUN) represent state 1, whereas the cation-bound NorM-NG (PDB ID code 4HUL) and NorM-VC structures (PDB ID codes 3MKU and 3MKT) emulate state 2 and state 3, respectively. TM1 and TM2 are simplified as a cyan stick, TM7 and TM8 as a thick yellow stick, and TM10 as a thin yellow stick.")
