Summary of Progress
The overall goals of this project are based on a multidisciplinary analysis of the different dynamic transitions that define gating event in K channels. These include relatively slow (ms) helix tilt and hinge-bending motions in the inner bundle gate, very slow (seconds) rearrangements in the selectivity filter (giving rise to C-type inactivation) and very fast (Flicker) transitions also associated with rearrangements at the selectivity filter. Within these wide ranging set of kinetics we plan, in the coming years, to examine the dynamics of ion conduction and the interplay between ion occupancy and selectivity filter conformation.
The types of questions we plan to respond can be approached only because of the robustness and experimental maturity of the system at hand (KcsA). Members of this Project have shown that KcsA represents an ideal system to study fundamental mechanisms of ion permeation and gating, and that these findings are directly applicable to eukaryotic systems. More recently, structure-function experiments led us to identify residues playing a critical role in C-type inactivation, flicker gating and pH sensitivity. Because of these findings, we are in a position to investigate the events linking the different conformations of the channel with the activation and inactivation processes at different time scales. We pursue a concerted approach using semi-synthesis of KcsA, patch clamp, X-ray diffraction and a host of spectroscopic methods (EPR, NMR, and 2D-IR) to develop, with the aid of cutting edge computational methods, an understanding of the conformational changes taking place at the selectivity filter when it conducts ions and when it inactivates.
Research Highlights
Click to find out more about each research highlight (RH).
with Benoît Roux
Changes in distances and distance cistributios at the outer versibule upon C-type inactivation. CW dipolar coupling in the Closed-Conductive.
Click to enlarge.An interesting paradox resulted from considering our C-type inactivated KcsA structure in the context of earlier functional studies that correlated conformational changes in the outer vestibule of K+ channels and the development of C-type inactivation. On one hand, metal bridge cysteine crosslinking engineering suggested relatively large changes at the external opening of the filter (Residues 448, 449 in Shaker).
The selectivity filter and surrounding structures play a crucial role as an inactivation gate in ion conduction. We have monitored the conformational changes in the outer vestibule of KcsA during gating using electrophysiological and EPR measurements to gain insight on the dynamic properties of these conformational fluctuations. pH-jump measurements shows that Cd2+ facilitates the rate of inactivation of outer vestibule mutant Y82C-KcsA, however, there is no effect of Cd2+ on the non-inactivating E71A/Y82C-KcsA. This suggests that the outer vestibule has different conformations in the inactivated and non-inactivated states of KcsA.
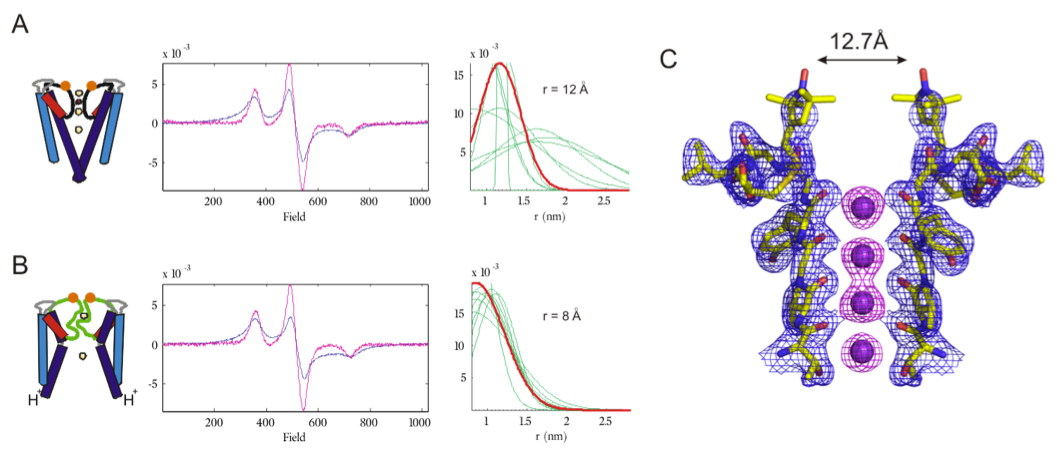
However, L81 and Y82, the equivalent residues in KcsA show little or no movement in our Open-Inactivated KcsA structures. We investigated this apparent paradox by monitoring the conformational changes in the outer vestibule of KcsA during gating using electrophysiology/Cd2+ bridges and EPR measurements to gain insight on the dynamic properties of these conformational fluctuations in KcsA. Just like in Shaker, pH-jump measurements showed that Cd2+ facilitates the rate of inactivation of outer vestibule mutant Y82C-KcsA, however, we found no effect of Cd2+ on the non-inactivating E71A/Y82C-KcsA. Distance measurements using CW-EPR at low temperatures show that the diagonal distance between spin labels bound to tandem-dimer Y82C and E71A/Y82C KcsA is ~12 Å when the lower gate is closed at pH7 (Figure, part A). This diagonal inter-spin in the closed state is in excellent agreement with the distance seen in the crystal structure of the spin label linked to Y82C KcsA in the closed state (Fig. part C). Interestingly, the distance between the spin labels bound to Y82C shortens to 8 Å upon inactivation (Fig. part B). Based on these results we suggest that local dynamics plays a major role in generating the inactivation-inducing Cd2+ bridges, as they do so by trapping the minimal inter-subunit distance between fluctuating residues in the external vestibule.

Spectroscopic analysis of the C-terminal domain conformational rearrangements. Structural rearrangements underlying channel opening.
Click to enlarge.
with Anthony Kossiakoff and Shohei Koide
Previously, we determined the crystal structure of full-length (FL) KcsA in its closed conformation using chaperone-assisted crystallography,. The FL structure reveals that the C-terminal domain (CTD) extends ~70 Å towards the cytoplasm as a canonical four helix bundle stabilizing the closed state. Electrophysiological analysis of KcsA/Fab4 complex demonstrates that CTD remains as a bundle during activation gating. This motivated us to crystallize open conformation (OC) of FL KcsA using the same antibody fragments and a recently developed constitutively open mutant, which was crystallized in its truncated form. This was carried out as a resul of our interactions with Core D3, to generate synthetic binders as crystallization chaperones.
We solved the FL KcsA/Fab2 complex structure in the open state at 3.9 Å resolution. FL open-structure bends at G104 and exhibits ~10 Å displacement at the V115 creating four side windows large enough to accommodate hydrated K+ ions right below the gate. The CTD remains as four helix bundle, exerting strain on the bulge helices which connects the bundle and the transmembrane domain.
Based on the current structure and an earlier set of truncated KcsA structures displaying different degree of openings, we suggest that the cytoplasmic domain not only stabilizes the closed state but also fine tunes the level of opening at the activation gate and thus determine the level of inactivation occurring at the selectivity filter. Brownian Dynamics, simulation, electrophysiological studies and electrostatic calculation are ongoing efforts to dissect the ion permeation pathway.

Kinetic effects of side chain substitutions at position 71.
Click to enlarge.Modal-gating shifts represent an effective regulatory mechanism by which ion channels control the extent and time course of ionic fluxes. Under steady-state conditions, the K+ channel KcsA displays three distinct gating modes, high-Po, low-Po and a high-frequency flicker mode, each with about an order of magnitude difference in their mean open times. In KcsA, the hydrogen bond network between Glu71, Asp80 and Trp67 that surrounds the selectivity filter has been shown to regulate C-type inactivation, with the Glu71-Asp80 pair having the strongest influence on selectivity filter stability. Over the past year, we have shown that in the absence of C-type inactivation, mutations at the pore-helix position Glu71 unmask a series of kinetically distinct modes of gating in a side-chain-specific way, which mirror those seen in wild-type channels. Results from high-resolution crystal structures along with molecular dynamic simulations suggest that specific interactions in the side-chain network surrounding the selectivity filter, in concert with ion occupancy, alter the relative stability of pre-existing conformational states of the pore. These findings highlight the key role of the selectivity filter in regulating modal gating behavior in K+ channels.
Interactions with Cores and Other Sections of the Consortium
The ongoing solution and solid state NMR experiments as well as the upcoming 2D-IR measurements are closely linked to the Chemical Synthesis Core D2 and the Protein Expression and Purification Core D1 for sample preparation, and the Computational Modeling Core D4 for protein dynamics. We expect a continued interaction with the Synthetic Antigen Binder and Crystallography Core D3 to generate novel Fabs to the extracellular regions of KcsA, aiming to generate new, higher resolution structures of KcsA gating intermediates. We will also continue tot exploit our recent success in crystallizing membrane proteins, using conformationally sensitive antibodie frangments. All the computational analysis continue to make extensive use of the spectral simulation tools (2D IR, NMR, ESR) and the transition pathway methodologies (String Method with swarm-of-trajectories) under development by the Computational Core D4.

