Proteins may undergo multiple conformational changes required for their function. One strategy used to estimate target site positions in unknown structural conformations involves single-pair resonance energy transfer (RET) distance measurements. However, interpretation of inter-residue distances is difficult when applied to 3D structural rearrangements, especially in homomeric systems. Probe diffusion further complicates this task by biasing measurements towards shorter distances. Lanthanide resonance energy transfer (LRET) is an ideal technique for simultaneously resolving multiple distances within a protein. Hyde et al. (2012) recently combined LRET with an ensemble of numerical methods to form the Symmetric Nano-Positioning System (SNPS), which allows accurate 3D positioning and inter-probe distance estimation in functional homomeric proteins.
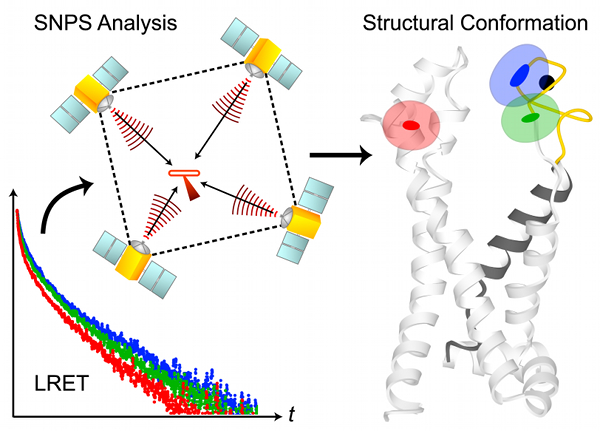
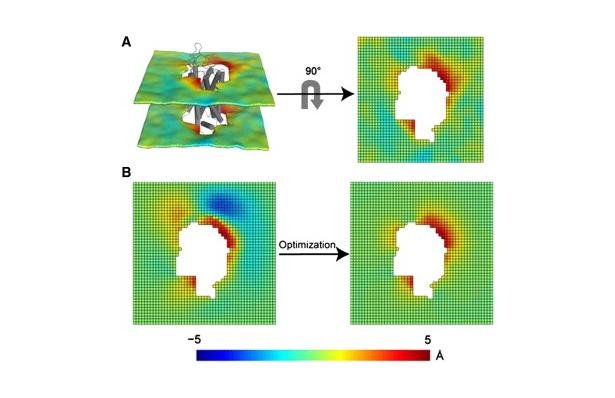
SNPS determines the 3D position of lanthanide donors [satellites] attached to a target site (one per subunit), relative to a single fluorescent acceptor [antenna] placed in a static reference site, as illustrated in the above figure. The acceptor’s position and accessible volume can be modeled from its structure-based labeling site by several methods, including dihedral scan analysis. SNPS can be applied to all defined conformational states of the protein and with simultaneous functional recordings. Satellite-to-antenna distances are encoded in time-resolved LRET lifetime decays. SNPS directly fits a 3D geometric model of satellite positions to LRET lifetime measurements using an inverse trilateration-based curve fitting procedure. Global analysis implementation fits the geometric model to an ensemble of replicate measurements. Numerical and analytical tools are integrated to account for probe diffusion and evaluate the confidence region of fitted positions. SNPS is well-suited to estimate 3D conformational changes at the target site between defined conformational states. In its first application, SNPS was used to determine the position of a functional voltage-gated potassium channel’s voltage sensor in its three major conformations [H. Clark Hyde, Walter Sandtner, Ernesto Vargas, Alper T. Dagcan, Janice L. Robertson, Benoit Roux, Ana M. Correa, Francisco Bezanilla (2012), Structure 20(10): 1629-1640].
The SNPS Toolbox contains the following stand-alone programs that implement the SNPS method:
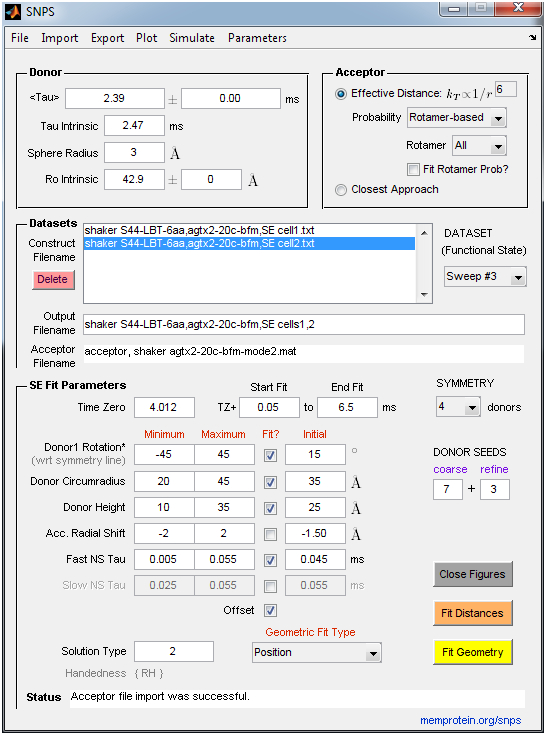
- SNPS: Performs inverse trilateration-based curve fitting of LRET lifetime decays to estimate the 3D position of lanthanide donors [satellites] attached to a target site (one per subunit), relative to a single fluorescent acceptor [antenna] placed in a static reference site. An acceptor file must be supplied by the user that specifies the acceptor’s coordinates and accessible volume. SNPS can alternatively fit all donor-acceptor distances constrained such that donors must lie in a polygon (symmetric) geometry, without any knowledge of acceptor position. SNPS can also simulate LRET lifetime decays at a user-defined donor position and noise level to explore the relation between donor position and decay shape. LRET lifetime decays are expected to be sensitized emission measured by a photomultiplier tube in analog mode (Poisson noise). The user imports lifetime decay files (ASCII .txt) and an acceptor file (ASCII .txt or Matlab .mat). See the “Example Data” folder for examples of required formatting. Fits are accompanied by a comprehensive set of output figures and a solution file.
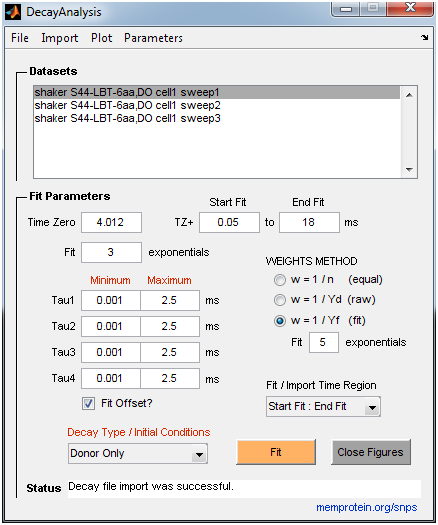
- DecayAnalysis: Performs model-free curve fitting of multi-exponential decays to estimate time constants and amplitudes (up to 4 exponential components). It is intended for donor-only time constant analysis of LRET experiments, but can also be used for general exponential fitting. The donor-only time constant is a required parameter for the SNPS program. The user can place constraints on time constants and define a weights scheme appropriate for the measurement uncertainty (e.g., Poisson vs. Gaussian noise). A large set of measurements can be fit quickly with basic statistical analysis reported. The user imports lifetime decay files in ASCII (.txt) format. See the “Example Data” folder for examples of required formatting.
SNPS and Decay Analysis Screen Guides
Download the software (version 2012.1):
Windows 64-bit: SNPS_Toolbox_win64.exe
Windows 32-bit: SNPS_Toolbox_win32.exe
Installation: Clicking on the downloadable .exe file will directly launch the Matlab Component Runtime (MCR) installation process and extract the SNPS and DecayAnalysis applications. This is a one-time installation, after which both applications can be launched. Installation of the included MCR is required even if you currently have Matlab or other MCR versions installed. Regarding processing speed, CPU speed takes precedence over the number of cores. The extracted folder also contains an “Example Data” folder with example data from the publication: AgTx2[II]-D20C-BODIPY-FL-maleimide acceptor cloud and Shaker S4(4) LBT construct sensitized emission (SE) and donor-only (DO) lifetime decays. Fit progress and information is displayed in an accompanying DOS window. Output files are written to a subfolder of the imported data directory.
The software was developed and written by H. Clark Hyde from Francisco Bezanilla’s group at the University of Chicago. Please contact us with questions or suggestions using the comments form below.