The Conformational Dynamics in the CLC Channel/Transporter Family Project of the Membrane Protein Structural Dynamics Consortium (MPSDC) has published its first publication titled “Water access points and hydration pathways in CLC H+/Cl- transporters” in Proceedings of the National Academy of Sciences of the United States of America (PNAS). This Consortium Project is spearheaded by Principal Investigator Merritt Maduke. The laboratories of Marc Baldus, Emad Tajkhorshid, and Hassane Mchaourab are also collaborators in the Project’s ongoing research.
Water access points and hydration pathways in CLC H+/Cl- transporters

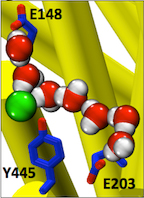
Figure 3. Entryways of water into the central hydrophobic region. (Han et al. 2013)
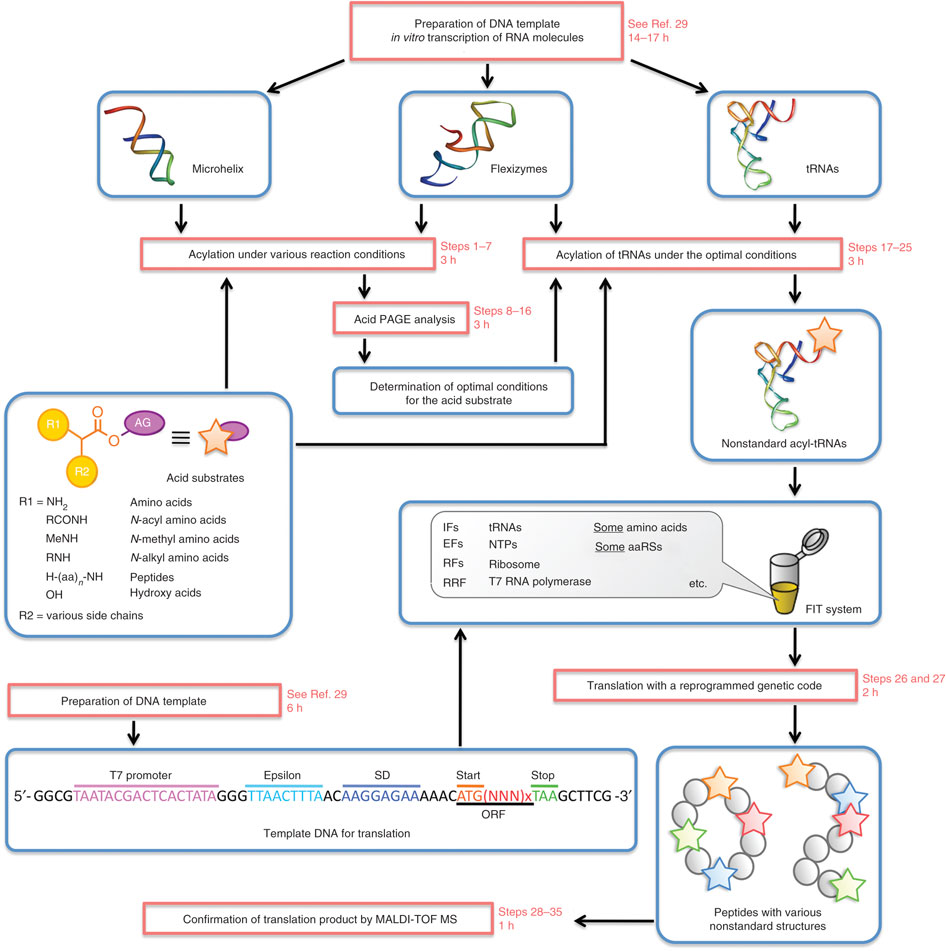
Abstract: CLC transporters catalyze transmembrane exchange of chloride for protons. Although a putative pathway for Cl− has been established, the pathway of H+ translocation remains obscure. Through a highly concerted computational and experimental approach, we characterize microscopic details essential to understanding H+-translocation. An extended (0.4 µs) equilibrium molecular dynamics simulation of membrane-embedded, dimeric ClC-ec1, a CLC from Escherichia coli, reveals transient but frequent hydration of the central hydrophobic region by water molecules from the intracellular bulk phase via the interface between the two subunits. We characterize a portal region lined by E202, E203, and A404 as the main gateway for hydration. Supporting this mechanism, site-specific mutagenesis experiments show that ClC-ec1 ion transport rates decrease as the size of the portal residue at position 404 is increased. Beyond the portal, water wires form spontaneously and repeatedly to span the 15-Å hydrophobic region between the two known H+ transport sites [E148 (Gluex) and E203 (Gluin)]. Our finding that the formation of these water wires requires the presence of Cl− explains the previously mystifying fact that Cl− occupancy correlates with the ability to transport protons. To further validate the idea that these water wires are central to the H+ transport mechanism, we identified I109 as the residue that exhibits the greatest conformational coupling to water wire formation and experimentally tested the effects of mutating this residue. The results, by providing a detailed microscopic view of the dynamics of water wire formation and confirming the involvement of specific protein residues, offer a mechanism for the coupled transport of H+ and Cl− ions in CLC transporters.
The results were published in Proceedings of the National Academy of Sciences of the United States of America, and are currently made available as an e-publication ahead of print. The citation is as follows:
Wei Han, Ricky C. Cheng, Merritt C. Maduke, and Emad Tajkhorshid.
Water access points and hydration pathways in CLC H+/Cl− transporters
PNAS 2013; published ahead of print December 30, 2013, doi:10.1073/pnas.1317890111
Learn more about this publication »
Significance
CLC transporters are biologically essential proteins that catalyze the transmembrane exchange of chloride for protons. The permeation pathway for chloride through the transporters has been well characterized. In this publication, Han et al. study the more elusive permeation pathway for protons. Through computational modeling, they show that water molecules can permeate deep inside the protein and form continuous wires. To test the hypothesis that these water wires mediate proton transport, they mutated residues predicted to impede water wire This research article reports results from the tightly coordinated efforts of a computational and an experimental lab brought together by the Consortium. The study addresses a critical question about the CLC transporter mechanism: how does H+ traverse the hydrophobic expanse of the CLC protein?formation and experimentally evaluated the effects of the mutations. The results from their concerted computational and experimental approach strongly support the role of water in proton transport by CLCs and provide a valuable framework for investigating their overall transport mechanism.
In a commentary piece published by PNAS this month, Mounir Tarek (National Center of Scientific Research at the University of Lorraine, France) describes the significance of this paper for future research of chloride channels, and highlights the fruitful combination of simulation and experimentation: “In PNAS, Han et al. used molecular dynamics (MD) simulations of the CLC-ecl, a CLC exchanger from Escherichia coli to specifically address this issue. The predictions of their calculations were tested by additional experiments, providing a robust description of the molecular prerequisites to proton transport in CLC-ecl and a framework for refining models of the Cl-/H+-coupled transport in CLCs.”
Indeed, this research article reports results from the tightly coordinated efforts of a computational and an experimental lab brought together by the Consortium. The study addresses a critical question about the CLC transporter mechanism: how does H+ traverse the hydrophobic expanse of the CLC protein? The MD simulations performed reveal water dynamics, water-wire formation, and side-chain conformational change not observed in any of the static crystal structures. The functional analyses validated predictions of the simulations and confirm the importance of water dynamics in the transport mechanism. The simulations further reveal that Cl- binding is critical for water-wire formation, thus providing a satisfying explanation for the puzzling experimental observation that Cl- occupancy correlates with the ability of CLCs to transport H+. These studies provide a crucial framework for understanding how H+ and Cl- binding and translocation steps are coordinated in the CLC transporters to control stoichiometric transport.
About the project
 The CLC family of chloride channels and transporters is necessary for proper neuronal, cardiovascular, and epithelial function. One of the important aspects of this family of transport proteins is that minute changes in their amino acid sequence can result in a shift in their operation mode from a channel to a transporter. Studying the structural dynamics of CLCs can therefore provide fundamental information on the nature of structural and dynamical differences between passive channels and active transporters.
The CLC family of chloride channels and transporters is necessary for proper neuronal, cardiovascular, and epithelial function. One of the important aspects of this family of transport proteins is that minute changes in their amino acid sequence can result in a shift in their operation mode from a channel to a transporter. Studying the structural dynamics of CLCs can therefore provide fundamental information on the nature of structural and dynamical differences between passive channels and active transporters.
The Conformational Dynamics in the CLC Channel/Transporter Family project addresses the multiple structural conformations that underlie the dual function of ClC- proteins as both channels and coupled transporters. Using a combination of NMR (solution and solid-state) and molecular dynamics simulations, the multiple conformations that support closely-coupled, stoichiometric ion transport will be accessed by binding and unbinding its two ligands, (Cl- and H+). Additional efforts are made made to use conformation-specific ligands to “lock” CLC proteins in order to study these conformations by crystallography, EPR, and NMR.
 Cartoon representation of T4 lysozyme (T4L) with 37 spin-labeled sites (colored in blue). As an example, the structural formula of two spin label side chains at positions 62 and 109 in T4L is shown here. The Restrained-Ensemble (RE) simulation method has been developed to restrain the calculated spin-spin distance distributions obtain from MD simulation to match with those obtained from the ESR/DEER, (B) From RE simulation, the dynamics of nitroxide oxygen of spin-labels around the Cα atom was obtained, (C) A simplified dummy nitroxide spin-label was designed and parameterized, (D) The simplified dummy spin-labels along with RE simulation refined distorted structures of T4L which is illustrated with root mean square deviation of the backbone atoms with respect to the X-ray structure (colored in blue). NOE-like distance restraints were unable to refine many of the distorted structures (colored in black).")
 Extracellular top view of NaVAb. In dark blue is the VSD and in dark red is the PD of one monomer. B) A side view of two of four monomers (removed to ease visualization) of NaVAb. The alpha carbon of Glutamate 177 is represented as a black sphere. (PDB ID 3RVY)")
 Two of four pore domains isolated from remaining structure. The two pore helices are indicated with arrows (P1 and P2) and the selectivity filter (SF). The alpha-carbon of glutamate 177 in the selectivity filter has been rendered as a black sphere to ease localization. B) A single voltage sensing domain of NaVAb is depicted in cartoon format with the four sensing charges (R1-R4) of the S4 helix depicted in stick format (dark red). (PDB ID 3RVY)")





