Biomolecular DNP- supported NMR spectroscopy using site directed spin labeling
Authors: Elwin A.W. van der Cruijsen, Eline J. Koers, Claire Sauvee, Raymond E. Hulse, Markus Weingarth, Olivier Ouari, Eduardo Perozo, Paul Tordo, and Marc Baldus.
In the press, Journal of the American Chemical Society. (Consortium project: Dynamics of Ion Permeation and Conformation Coupling in KcsA)
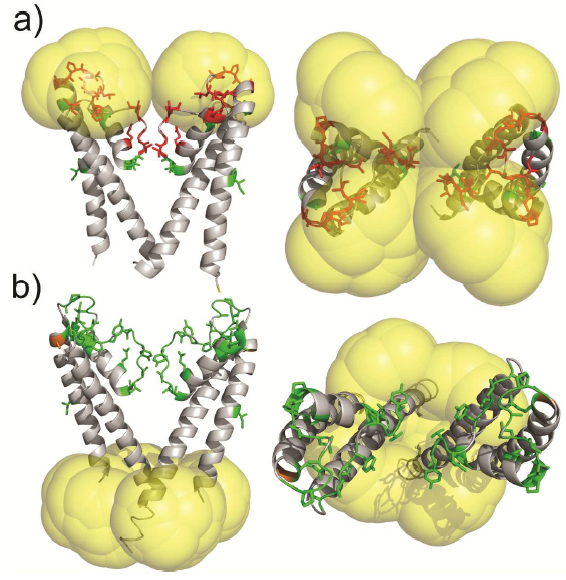
Molecular probes that combine the benefits of enhanced spectroscopic sensitivity with site-specific localization have significantly expanded our ability to track molecular structure and function in applications ranging from cell biology to material science . In the field of magnetic resonance, dynamic nuclear polarization (DNP) has become a widely usable method to significantly enhance overall spectroscopic sensitivity in NMR and MRI. Here, the authors show that DNP can be established by creating local spin clusters via site-directed spin labeling using mono- or biradicals. Applied to a membrane-embedded potassium channel, we show that this approach can significantly enhance NMR sensitivity while preserving the intrinsic spectroscopic properties of (bi)radicals as paramagnetic relaxation enhancers.
The results suggest that the creation of local spin clusters can generate sizable DNP enhancements while preserving the intrinsic benefits of PRE-based NMR approaches. Our results are consistent with the idea that the magnitude in DNP enhancement are highly dependent on the nearest neighbor electron-electron distances.
13C NMR detects conformational change in the 100-kD membrane transporter Cl-C-ec1
Authors: Sherwin J. Abraham, Ricky C. Cheng, Thomas A. Chew, Chandra M. Khantwal, Corey W. Liu, Shimei Gong, Robert K. Nakamoto, and Merritt Maduke.
J Biomol NMR. 2015 Jan 29. (Consortium project: Conformational Dynamics in the CLC Channel Transporter Family. Consortium core: Membrane Protein Production)
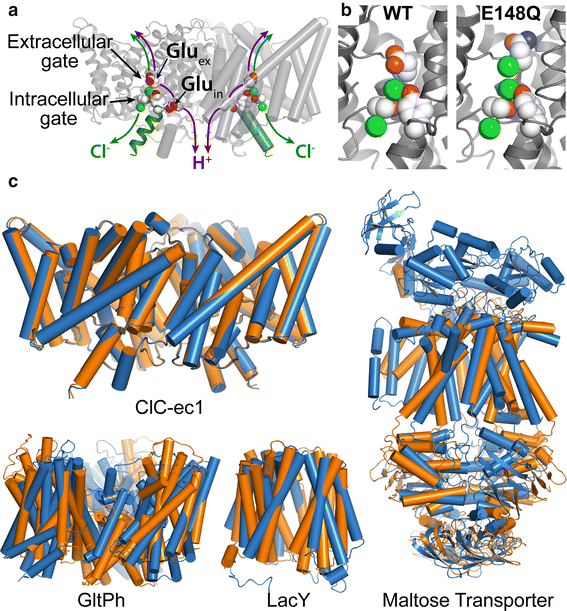
Members of the Cl-C (‘‘Chloride-Channel’’) family play central roles in cardiovascular, neuronal, bone, and epithelial function. Cl-C transporters catalyze the exchange of Cl--for H+ across cellular membranes. To do so, they must couple Cl-- and H+ binding and unbinding to protein conformational change. However, the sole conformational changes distinguished crystallographically are small movements of a glutamate side chain that locally gates the ion transport pathways. Therefore, our understanding of whether and how global protein dynamics contribute to the exchange mechanism has been severely limited. To overcome the limitations of crystallography, the authors used solution-state 13Cmethyl NMR with labels on methionine, lysine, and engineered cysteine residues to investigate substrate (H+) dependent conformational change outside the restraints of crystallization. They show that methyl labels in several regions report H+-dependent spectral changes. They identify one of these regions as Helix R, a helix that extends from the center of the protein, where it forms the part of the inner gate to the Cl-–permeation pathway, to the extracellular solution. The H+-dependent spectral change does not occur when a label is positioned just beyond Helix R, on the unstructured C-terminus of the protein.
Together, the results suggest that H+ binding is mechanistically coupled to Cl-osing of the intracellular access-pathway for Cl--. These studies set the stage for investigating the structural details and dynamics of this change.
Room-Temperature Distance Measurements of Immobilized Spin-Labeled Protein by DEER/PELDOR
Authors: Virginia Meyer, Michael A. Swanson, Laura J. Clouston, Przemyslaw J. Boratynski, Richard A. Stein, Hassane S. Mchaourab, Andrzej Rajca, Sandra S. Eaton, and Gareth R. Eaton.
Biophys J. 2015 Mar 10;108(5):1213-9. (Consortium Core: Membrane Protein Production)
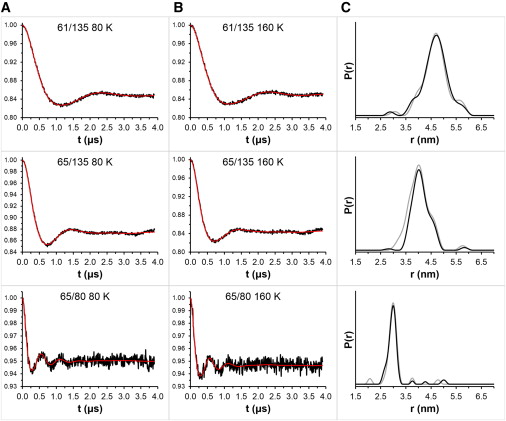
Nitroxide spin labels are used for double electron-electron resonance (DEER) measurements of distances between sites in biomolecules. Rotation of gem-dimethyls in commonly used nitroxides causes spin echo dephasing times (Tm) to be too short to perform DEER measurements at temperatures between ~80 and 295 K, even in immobilized samples. A spirocyclohexyl spin label has been prepared that has longer Tm between 80 and 295 K in immobilized samples than conventional labels. Two of the spirocyclohexyl labels were attached to sites on T4 lysozyme introduced by site-directed spin labeling. Interspin distances up to ~4 nm were measured by DEER at temperatures up to 160 K in water/glycerol glasses. In a glassy trehalose matrix the Tm for the doubly labeled T4 lysozyme was long enough to measure an interspin distance of 3.2 nm at 295 K, which could not be measured for the same protein labeled with the conventional 1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-(methyl) methanethio-sulfonate label.
The leap from 80 K DEER measurements to room temperature is an important step toward DEER measurement in physiological environments. The current fundamental requirement for DEER of protein immobilization provides additional avenues toward improvement of the technique.
Architecture of the nuclear pore complex coat
Authors: Tobias Stuwe, Ana R. Correia, Daniel H. Lin, Marcin Paduch, Vincent T. Lu, Anthony A. Kossiakoff, and André Hoelz.
Science. 2015 Mar 6;347(6226):1148-52. (Consortium core: Synthetic Antigen Binder (SAB) Generation and Crystallography)
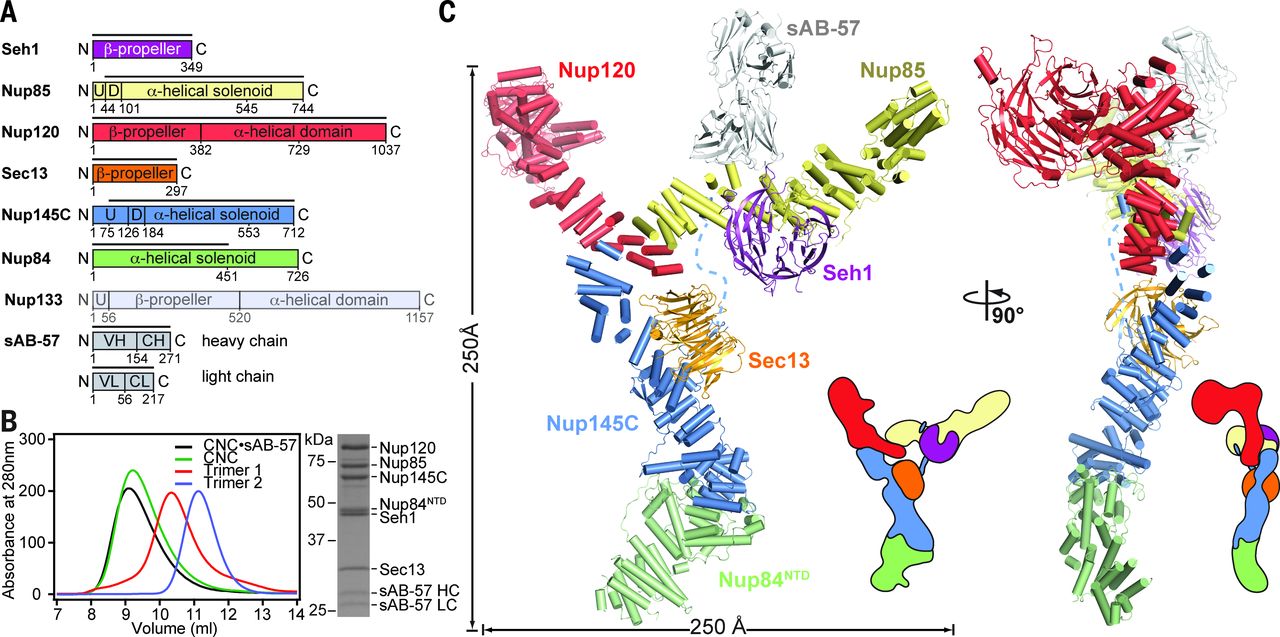
The nuclear pore complex (NPC) constitutes the sole gateway for bidirectional nucleocytoplasmic transport. Despite half a century of structural characterization, the architecture of the NPC remains unknown. In this research report, the authors present the crystal structure of a reconstituted ~400-kilodalton coat nucleoporin complex (CNC) from Saccharomyces cerevisiae at a 7.4 angstrom resolution. The crystal structure revealed a curved Y-shaped architecture and the molecular details of the coat nucleoporin interactions forming the central “triskelion” of the Y. A structural comparison of the yeast CNC with an electron microscopy reconstruction of its human counterpart suggested the evolutionary conservation of the elucidated architecture. Moreover, 32 copies of the CNC crystal structure docked readily into a cryoelectron tomographic reconstruction of the fully assembled human NPC, thereby accounting for ~16 megadalton of its mass.
Multilevel Summation Method for Electrostatic Force Evaluation
Authors: David J. Hardy, Zhe Wu, James C. Phillips, John E. Stone, Robert D. Skeel, and Klaus Schulten.
J Chem Theory Comput. 2015 Feb 10;11(2):766-779. PMCID: PMC4325600. (Consortium core: Computational Modeling Core)

Significant long-range electrostatic interactions arise in many biomolecular systems, such as negatively charged DNA and RNA, polar or charged membranes, ion channels, and electrostatic steering of protein−protein and enzyme−substrate association. Accordingly, electrostatic interactions need to be accurately represented in molecular modeling calculations. The computational cost increases in principle as N2, where N is the number of charged partiCl-es in the system.
The multilevel summation method (MSM) offers an efficient algorithm utilizing convolution for evaluating long-range forces arising in molecular dynamics simulations. Shifting the balance of computation and communication, MSM provides key advantages over the ubiquitous partiCl-e−mesh Ewald (PME) method, offering better scaling on parallel computers and permitting more modeling flexibility, with support for periodic systems as does PME but also for semiperiodic and nonperiodic systems. The version of MSM available in the simulation program NAMD is described, and its performance and accuracy are compared with the PME method. The accuracy feasible for MSM in practical applications reproduces PME results for water property calculations of density, diffusion constant, dielectric constant, surface tension, radial distribution function, and distance dependent Kirkwood factor, even though the numerical accuracy of PME is higher than that of MSM. Excellent agreement between MSM and PME is found also for interface potentials of air−water and membrane−water interfaces, where long-range Coulombic interactions are crucial. Applications demonstrate also the suitability of MSM for systems with semiperiodic and nonperiodic boundaries. For this purpose, simulations have been performed with periodic boundaries along directions parallel to a membrane surface but not along the surface normal, yielding membrane pore formation induced by an imbalance of charge across the membrane. Using a similar semiperiodic boundary condition, ion conduction through a graphene nanopore driven by an ion gradient has been simulated. Furthermore, proteins have been simulated inside a single spherical water droplet. Finally, parallel scalability results show the ability of MSM to outperform PME when scaling a system of modest size (less than 100 K atoms) to over a thousand processors, demonstrating the suitability of MSM for large-scale parallel simulation.
Ongoing is the development of improved interpolation for MSM to provide higher accuracy for a given polynomial degree p without increasing the computational cost. Future work inCl-udes also the calculation of dispersion forces without truncation with MSM-based NAMD; these forces, in particular, their long-range contribution, are considered to be important for membrane properties. With support in NAMD also for long-range dispersion forces, the present CHARMM-prescribed 12 Å cutoff/splitting distance can be used as a true control for MSM accuracy. High performance simulations will then be able to achieve practical accuracy with a reduced splitting distance, where a splitting distance of between 8 and 9 Å is expected to double the overall simulation performance.
Potential Application of Alchemical Free Energy Simulations to Discriminate GPCR Ligand Efficacy
Authors: Hui Sun Lee, Chaok Seok, and Wonpil Im.
J. Chem. Theory Comput. 2015 Feb 10;11:1255-1266. (Consortium core: Computational Modeling Core)
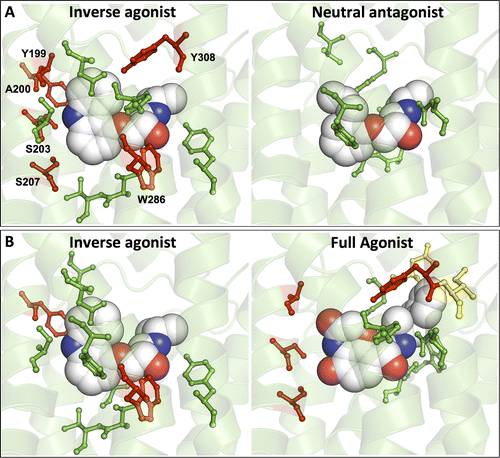
G protein-coupled receptors (GPCRs) constitute the largest protein superfamily in the human genome with almost 1,000 members. They play key functional roles as major contributors of information flow from the outside to the inside of the cell, making them one of the most important protein families. As a result of their broad influence on human physiology and behavior, GPCRs are arguably the most promising targets for development of new and more effective therapeutic agents.
Based on the fact that GPCR-mediated signaling is modulated in a ligand-specific manner such as agonist, inverse agonist, and neutral antagonist (termed ligand efficacy), quantitative characterization of the ligand efficacy is essential for rational design of selective modulators for GPCR targets. As experimental approaches for this purpose are time-, cost-, and labor-intensive, computational tools that can systematically predict GPCR ligand efficacy can have a big impact on GPCR drug design. Here, the authors have performed free energy perturbation molecular dynamics simulations to calculate absolute binding free energy of an inverse agonist, a neutral antagonist, and an agonist to β2-adrenergic receptor (β2-AR) active and inactive states, respectively, in explicit lipid bilayers. Relatively short alchemical free energy calculations reveal that both the time series of the total binding free energy and decomposed energy contributions can be used as relevant physical properties to discriminate β2-AR ligand efficacy. This study illustrates a merit of the current approach over simple, fast docking calculations or highly expensive millisecond-time scale simulations.
It is the authors’ hope that their computational approach improves research and development efficiency in designing novel lead compounds targeting various GPCRs for the treatment of various human diseases.





