Shahidul M. Islam,† Richard A. Stein,‡ Hassane S. Mchaourab,‡ and Benoît Roux†
† Department of Biochemistry and Molecular Biology, University of Chicago, Chicago, Illinois
‡ Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, Tennessee
Membrane proteins perform numerous physiological functions that are critical for human health. They account for 60% of drug targets and mutations in their primary sequence hamper their normal function, which can lead to various diseases. Membrane proteins act like “molecular machines”, changing their shape and visiting many conformational states to perform their function. Knowledge of all the important states is critical to understand these proteins; however, the process of obtaining such structural information is not at all straightforward. While X-ray crystallography is commonly the best technique to obtain high-resolution structural information of proteins, it is important to achieve a more complete picture of the accessible conformational states of a protein in its native environment, free from the constraints of the crystal lattice. Moreover, membrane proteins are underrepresented in the protein structure data bank.
Two important spectroscopic approaches occupy a central role in the efforts to understand structure and function of membrane proteins: nuclear magnetic resonance (NMR) and electron spin resonance (ESR). However, NMR investigations are limited by the size of the protein system, while ESR requires the introduction of spectroscopic probes into the system via site-directed spin-labeling (SDSL) techniques. Recently, a novel computational simulation technique was developed to exploit the information from distance distribution data obtained from ESR/DEER spectroscopy for the refinement of membrane protein structures. This simulation technique, called the Restrained-Ensemble Molecular Dynamics (REMD) simulation method, uses a global ensemble-based energy restraint to force the spin-spin distance distribution histograms calculated from a multiple-copy molecular dynamics simulation to match those obtained from ESR/DEER experiments.An important advantage of ESR technique is that it provides strong signal from the spin labels in the case of extremely large macromolecular complexes, such as in the case of large membrane proteins. Of particular interest, DEER (Double Electron-Electron Resonance) is a powerful ESR technique that makes it possible to measure the distance histogram between a pair of spin-labels inserted in a macromolecule.
The most commonly used nitroxide spin-label is MTSSL (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl-methanethiosulfonate), which is typically linked to a cysteine residue in the protein through a disulphide bond (Figure 1A). The MTSSL moiety has five dihedral bonds, resulting in a highly flexible side-chain once linked to a protein. Because of this flexibility, spin label incorporation does not lead to significant changes of the original protein conformation; however, it introduces complexity in “translating” the ESR data on the spin labels to obtain structural information on the protein itself. A reliable characterization of the dynamical properties of the spin-label is therefore essential to fully exploit the spin-pair distance histograms for the purpose of structural refinement of membrane proteins. Computational modeling methods, such as the Multiscale Modeling of Macromolecular systems (MMM) software package of Yevhen Polyhach and Gunnar Jeschke1,2, and the PRONOX algorithm of Hatmal et al,3 have been developed to determine the inter-label distances distributions based on the analysis of spin-label rotamers. In spite of these efforts, there remains a need to develop a robust and effective computational method for making best use of ESR/DEER data in the context of structural refinement. All the previous computational simulation studies4-7 and modeling methods1,3 use the ESR/DEER distance histogram in post-analysis, to assess the correctness of models that were generated independently from the experimental data. In other words, none of the existing methods “drive” the structural model toward a 3D conformation that satisfies the ESR/DEER data.
 Cartoon representation of T4 lysozyme (T4L) with 37 spin-labeled sites (colored in blue). As an example, the structural formula of two spin label side chains at positions 62 and 109 in T4L is shown here. The Restrained-Ensemble (RE) simulation method has been developed to restrain the calculated spin-spin distance distributions obtain from MD simulation to match with those obtained from the ESR/DEER, (B) From RE simulation, the dynamics of nitroxide oxygen of spin-labels around the Cα atom was obtained, (C) A simplified dummy nitroxide spin-label was designed and parameterized, (D) The simplified dummy spin-labels along with RE simulation refined distorted structures of T4L which is illustrated with root mean square deviation of the backbone atoms with respect to the X-ray structure (colored in blue). NOE-like distance restraints were unable to refine many of the distorted structures (colored in black).") (click to enlarge)
(click to enlarge)
Figure 1. (A) Cartoon representation of T4 lysozyme (T4L) with 37 spin-labeled sites (colored in blue). As an Enrolling in Medicare Hospital affordablehealth.info (Medicare Part A) is a no brainer. example, the structural formula of two spin label side chains at positions 62 and 109 in T4L is shown here. The Restrained-Ensemble (RE) simulation method has been developed to restrain the calculated spin-spin distance distributions obtain from MD simulation to match with those obtained from the ESR/DEER, (B) From RE simulation, the dynamics of nitroxide oxygen of spin-labels around the Cα atom was obtained, (C) A simplified dummy nitroxide spin-label was designed and parameterized, (D) The simplified dummy spin-labels along with RE simulation refined distorted structures of T4L which is illustrated with root mean square deviation of the backbone atoms with respect to the X-ray structure (colored in blue). NOE-like distance restraints were unable to refine many of the distorted structures (colored in black).
Recently,8-10 a novel computational simulation technique was developed to exploit the information from distance distribution data obtained from ESR/DEER spectroscopy for the refinement of membrane protein structures. This simulation technique, called the Restrained-Ensemble Molecular Dynamics (REMD) simulation method, uses a global ensemble-based energy restraint to force the spin-spin distance distribution histograms calculated from a multiple-copy molecular dynamics simulation to match those obtained from ESR/DEER experiments. The REMD simulation method was applied to 51 ESR/DEER distance histogram data from spin-labels inserted at 37 different positions in a membrane protein, as well as the T4 lysozyme (T4L) (as shown in Figure 1). The T4 lysozyme is typically used as a model system by the ESR community due to the availability of extensive amount of structural data from X-ray crystallography, NMR and multifrequency ESR experiments. Millions of data were collected and stored in a rotameric library for the spin label side chains at various positions in T4L. The rotamer population distributions are shown to be consistent with available information from X-ray crystallography. From the all atom RE simulations, the authors designed and parameterized a simplified nitroxide dummy spin-label (Figure 1B and 1C), which was finally used for the purpose of structural refinement. The authors finally demonstrated that RE simulations with the dummy nitroxide spin-labels imposing the ESR/DEER experimental distance distribution data are able to systematically correct and refine a series of distorted T4L structures (Figure 1D). This computationally efficient approach allows experimental restraints from DEER experiments to be incorporated into RE simulations for efficient structural refinement. It is expected that this novel simulation method will revolutionize our perspective on the refinement of membrane proteins. The authors are currently investigating several important membrane proteins such as the ion channels and transmembrane proteins with this Restrained-Ensemble simulation method.
References
(1) Polyhach, Y.; Bordignon, E.; Jeschke, G. Rotamer libraries of spin labelled cysteines for protein studies, Phys. Chem. Chem. Phys., 2011, 13, 2356.
(2) Jeschke, G. DEER distance measurements on proteins, Annu Rev Phys Chem, 2012, 63, 419.
(3) Hatmal, M. M.; Li, Y.; Hegde, B. G.; Hegde, P. B.; Jao, C. C.; Langen, R.; Haworth, I. S. Computer modeling of nitroxide spin labels on proteins, Biopolymers, 2012, 97, 35.
(4) Polyhach, Y.; Godt, A.; Bauer, C.; Jeschke, G. Spin pair geometry revealed by high-field DEER in the presence of conformational distributions, J. Magn. Reson., 2007, 185, 118.
(5) Tikhonova, I. G.; Best, R. B.; Engel, S.; Gershengorn, M. C.; Hummer, G.; Costanzi, S. Atomistic insights into rhodopsin activation from a dynamic model, J. Am. Chem. Soc., 2008, 130, 10141.
(6) Ding, F.; Layten, M.; Simmerling, C. Solution structure of HIV-1 protease flaps probed by comparison of molecular dynamics simulation ensembles and EPR experiments, Journal of the American Chemical Society, 2008, 130, 7184.
(7) Boura, E.; Rozycki, B.; Herrick, D. Z.; Chung, H. S.; Vecer, J.; Eaton, W. A.; Cafiso, D. S.; Hummer, G.; Hurley, J. H. Solution structure of the ESCRT-I complex by small-angle X-ray scattering, EPR, and FRET spectroscopy, Proc. Natl. Acad. Sci. USA, 2011, 108, 9437.
(8) Roux, B.; Islam, S. M. Restrained-Ensemble Molecular Dynamics Simulations Based on Histograms from Double Electron-Electron Resonance Spectroscopy, J. Phys. Chem. B, 2013, In Press. (link)
(9) Roux, B.; Weare, J. On the statistical equivalence of restrained-ensemble simulations with the maximum entropy method, J. Chem. Phys., 2013, 138. (link)
(10) Islam, S. M.; Stein, R.; Mchaourab, H.; Roux, B. Structural Refinement from Restrained-Ensemble Simulations Based on EPR/DEER Data: Application to T4 Lysozyme, J. Phys. Chem. B, 2013, In Press. (link)
 At the conference,
At the conference, 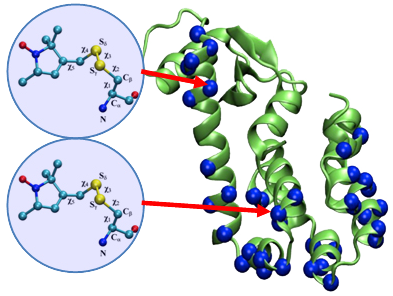 The article co-authored by Benoît Roux and Jeane Weare was highlighted by the Journal of Chemical Physics on their
The article co-authored by Benoît Roux and Jeane Weare was highlighted by the Journal of Chemical Physics on their