MPSDC Team Member Robert Nakamoto, who leads the Consortium’s Protein Production/Expression Core, is this year’s Chair of the 2014 Biophysical Society Program Committee. Nakamoto was interviewed several times by Biophysical Society TV for the occasion of the 2014 Biophysical Society meetings. You can check out the interviews below:
Interview with Francisco Bezanilla, MPSDC collaborator and President of the Biophysical Society
Biophysical Society president and MPSDC Team Member Francisco Bezanilla was interviewed today by Biophysical Society TV. You can check out the interview below:
Frontiers in Membrane Structural Protein Dynamics 2014 poster made available
Help us advertise Frontiers in Membrane Protein Structural Dynamics 2014 by circulating our newly designed meeting poster!

If you wish to print this poster, here are some pdf files that have been optimized for high quality printing:
- Ledger (11″ x 17″) size: http://memprotein.org/site-media/frontiers-2014-ledger.pdf
- A3 (11.7″ x 16.5″) size: http://memprotein.org/site-media/frontiers-2014-a3.pdf
- Letter (8.5″ x 11″) size: http://memprotein.org/site-media/frontiers-2014-letter.pdf
If you wish to publish this poster on the web or via email, here are some image links for embedding:
- http://memprotein.org/site-media/frontiers-2014-large.png
- http://memprotein.org/site-media/frontiers-2014-medium.png
- http://memprotein.org/site-media/frontiers-2014-small.png
- http://memprotein.org/site-media/frontiers-2014-thumbnail.png
Remember, please register early, as all of our previous conferences have been booked over capacity!
CLC Channel/Transporter Family Project team combines computational and experimental methodologies in PNAS publication
The Conformational Dynamics in the CLC Channel/Transporter Family Project of the Membrane Protein Structural Dynamics Consortium (MPSDC) has published its first publication titled “Water access points and hydration pathways in CLC H+/Cl- transporters” in Proceedings of the National Academy of Sciences of the United States of America (PNAS). This Consortium Project is spearheaded by Principal Investigator Merritt Maduke. The laboratories of Marc Baldus, Emad Tajkhorshid, and Hassane Mchaourab are also collaborators in the Project’s ongoing research.
Water access points and hydration pathways in CLC H+/Cl- transporters

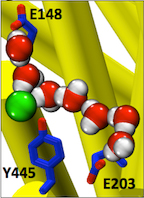
Figure 3. Entryways of water into the central hydrophobic region. (Han et al. 2013)
Abstract: CLC transporters catalyze transmembrane exchange of chloride for protons. Although a putative pathway for Cl− has been established, the pathway of H+ translocation remains obscure. Through a highly concerted computational and experimental approach, we characterize microscopic details essential to understanding H+-translocation. An extended (0.4 µs) equilibrium molecular dynamics simulation of membrane-embedded, dimeric ClC-ec1, a CLC from Escherichia coli, reveals transient but frequent hydration of the central hydrophobic region by water molecules from the intracellular bulk phase via the interface between the two subunits. We characterize a portal region lined by E202, E203, and A404 as the main gateway for hydration. Supporting this mechanism, site-specific mutagenesis experiments show that ClC-ec1 ion transport rates decrease as the size of the portal residue at position 404 is increased. Beyond the portal, water wires form spontaneously and repeatedly to span the 15-Å hydrophobic region between the two known H+ transport sites [E148 (Gluex) and E203 (Gluin)]. Our finding that the formation of these water wires requires the presence of Cl− explains the previously mystifying fact that Cl− occupancy correlates with the ability to transport protons. To further validate the idea that these water wires are central to the H+ transport mechanism, we identified I109 as the residue that exhibits the greatest conformational coupling to water wire formation and experimentally tested the effects of mutating this residue. The results, by providing a detailed microscopic view of the dynamics of water wire formation and confirming the involvement of specific protein residues, offer a mechanism for the coupled transport of H+ and Cl− ions in CLC transporters.
The results were published in Proceedings of the National Academy of Sciences of the United States of America, and are currently made available as an e-publication ahead of print. The citation is as follows:
Wei Han, Ricky C. Cheng, Merritt C. Maduke, and Emad Tajkhorshid.
Water access points and hydration pathways in CLC H+/Cl− transporters
PNAS 2013; published ahead of print December 30, 2013, doi:10.1073/pnas.1317890111
Learn more about this publication »
Significance
CLC transporters are biologically essential proteins that catalyze the transmembrane exchange of chloride for protons. The permeation pathway for chloride through the transporters has been well characterized. In this publication, Han et al. study the more elusive permeation pathway for protons. Through computational modeling, they show that water molecules can permeate deep inside the protein and form continuous wires. To test the hypothesis that these water wires mediate proton transport, they mutated residues predicted to impede water wire This research article reports results from the tightly coordinated efforts of a computational and an experimental lab brought together by the Consortium. The study addresses a critical question about the CLC transporter mechanism: how does H+ traverse the hydrophobic expanse of the CLC protein?formation and experimentally evaluated the effects of the mutations. The results from their concerted computational and experimental approach strongly support the role of water in proton transport by CLCs and provide a valuable framework for investigating their overall transport mechanism.
In a commentary piece published by PNAS this month, Mounir Tarek (National Center of Scientific Research at the University of Lorraine, France) describes the significance of this paper for future research of chloride channels, and highlights the fruitful combination of simulation and experimentation: “In PNAS, Han et al. used molecular dynamics (MD) simulations of the CLC-ecl, a CLC exchanger from Escherichia coli to specifically address this issue. The predictions of their calculations were tested by additional experiments, providing a robust description of the molecular prerequisites to proton transport in CLC-ecl and a framework for refining models of the Cl-/H+-coupled transport in CLCs.”
Indeed, this research article reports results from the tightly coordinated efforts of a computational and an experimental lab brought together by the Consortium. The study addresses a critical question about the CLC transporter mechanism: how does H+ traverse the hydrophobic expanse of the CLC protein? The MD simulations performed reveal water dynamics, water-wire formation, and side-chain conformational change not observed in any of the static crystal structures. The functional analyses validated predictions of the simulations and confirm the importance of water dynamics in the transport mechanism. The simulations further reveal that Cl- binding is critical for water-wire formation, thus providing a satisfying explanation for the puzzling experimental observation that Cl- occupancy correlates with the ability of CLCs to transport H+. These studies provide a crucial framework for understanding how H+ and Cl- binding and translocation steps are coordinated in the CLC transporters to control stoichiometric transport.
About the project
 The CLC family of chloride channels and transporters is necessary for proper neuronal, cardiovascular, and epithelial function. One of the important aspects of this family of transport proteins is that minute changes in their amino acid sequence can result in a shift in their operation mode from a channel to a transporter. Studying the structural dynamics of CLCs can therefore provide fundamental information on the nature of structural and dynamical differences between passive channels and active transporters.
The CLC family of chloride channels and transporters is necessary for proper neuronal, cardiovascular, and epithelial function. One of the important aspects of this family of transport proteins is that minute changes in their amino acid sequence can result in a shift in their operation mode from a channel to a transporter. Studying the structural dynamics of CLCs can therefore provide fundamental information on the nature of structural and dynamical differences between passive channels and active transporters.
The Conformational Dynamics in the CLC Channel/Transporter Family project addresses the multiple structural conformations that underlie the dual function of ClC- proteins as both channels and coupled transporters. Using a combination of NMR (solution and solid-state) and molecular dynamics simulations, the multiple conformations that support closely-coupled, stoichiometric ion transport will be accessed by binding and unbinding its two ligands, (Cl- and H+). Additional efforts are made made to use conformation-specific ligands to “lock” CLC proteins in order to study these conformations by crystallography, EPR, and NMR.
Publication by Schulten and Tajkhorshid featured as cover page for Journal of Computational Chemistry
Membrane Protein Structural Dynamics Consortium (MPSDC) team members Klaus Schulten and Emad Tajkhorshid from the Computational Modeling Core recently collaborated on an publication about a new Force Field Toolkit (ffTK), which minimizes common barriers to ligand parameterization through algorithm and method development, automation of tedious and error-prone tasks, and graphical user interface design. Distributed as a VMD plugin, ffTK facilitates the traversal of a clear and organized workflow resulting in a complete set of CHARMM-compatible parameters.
The article, titled Rapid parameterization of small molecules using the Force Field Toolkit was published in the Journal of Computational Chemistry and featured as a Cover Article for Volume 34, Issue 32. The journal provided the following caption along with the cover: “The Force Field Toolkit (ffTK), a new plugin for visual molecular dynamics by Christopher G. Mayne et al. on page 2757, aids users in the development of CHARMM/CGenFF-compatible force field parameters for small molecules. The primary function of ffTK is to generate quantum mechanical target data and optimize molecular mechanics force field parameters. The cover shows water interation profiles (center left), which are computed at each iteration of the partial atomic charge optimization, and torsion scans (left to right), which are used to compute potential energy surfaces during dihedral parameter optimization. ffTK also provides a suite of analytical tools to assess optimization metrics and parameter performance using embedded plotting utilities (background).”
Click the image to view the cover, and the inset text, in more detail:

- Advances in the Field
- Annual Meeting and Conference
- Behind the Scenes
- Events
- Ideas and Concepts
- In the Press
- News
- New Software
- 2019 Biological CryoEM workshop video and photos released
- 2019 Workshop on Advances and Challenges of Biological CryoEM
- Third Coast Workshop on Biological Cryo-EM announced for May 19, 2018
- Final Program for III Frontiers in Membrane Protein Structural Dynamics Announced
- Announcing Frontiers in Membrane Protein Structural Dynamics 2017
- July 2019 (1)
- March 2019 (1)
- March 2018 (1)
- November 2017 (1)
- June 2017 (1)
- February 2017 (2)
- January 2017 (1)
- November 2016 (1)
- May 2016 (2)
- October 2015 (2)
- September 2015 (2)
- August 2015 (1)
- May 2015 (1)
- April 2015 (1)
- March 2015 (4)
- February 2015 (1)
- January 2015 (1)
- October 2014 (2)
- June 2014 (1)
- May 2014 (4)
- April 2014 (7)
- February 2014 (4)
- January 2014 (3)
- December 2013 (1)
- November 2013 (2)
- October 2013 (1)
- September 2013 (2)
- August 2013 (1)
- May 2013 (3)
- April 2013 (3)
- March 2013 (2)
- November 2012 (1)
- October 2012 (2)
- June 2012 (2)
- April 2012 (3)
- March 2012 (4)
- February 2012 (2)
- January 2012 (4)
- December 2011 (1)
- November 2011 (3)
- October 2011 (3)
- September 2011 (1)
- July 2011 (5)
- November 2010 (1)
- September 2010 (1)
- August 2010 (2)
- Dr Garau on New Webserver: ANMPathway determines the most probable (lowest energy) transition pathway between two stable endpoints of a conformational transition
- Dr Garau on New Webserver: ANMPathway determines the most probable (lowest energy) transition pathway between two stable endpoints of a conformational transition
- Ashfaq Ahmad on ABC Transporter paper by Tajkhorshid laboratory recommended on F1000Prime
- admin on New Webserver: ANMPathway determines the most probable (lowest energy) transition pathway between two stable endpoints of a conformational transition
- Karen on New Webserver: ANMPathway determines the most probable (lowest energy) transition pathway between two stable endpoints of a conformational transition





